



















 Медицина
МедицинаПохожие презентации:



")


")
")


Валидация аналитических методик. Вебинар 6
1.
ВЕБИНАР 6ВАЛИДАЦИЯ
АНАЛИТИЧЕСКИХ МЕТОДИК
Анна Трифонова
25 декабря 2020 г.
2.
ПЛАН1
• Валидация
аналитических
методик
2
• Метрологические
характеристики в
валидации
3
• Базовые понятия
статистики в
валидации
Вебинар 6
Валидация
аналитических
методик
3.
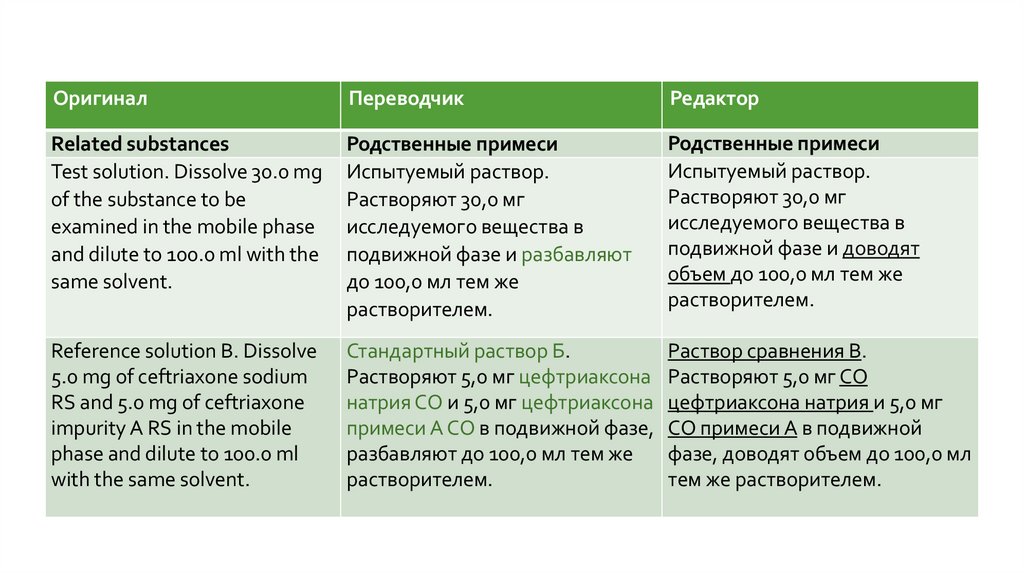
ОригиналПереводчик
Редактор
Related substances
Test solution. Dissolve 30.0 mg
of the substance to be
examined in the mobile phase
and dilute to 100.0 ml with the
same solvent.
Родственные примеси
Испытуемый раствор.
Растворяют 30,0 мг
исследуемого вещества в
подвижной фазе и разбавляют
до 100,0 мл тем же
растворителем.
Родственные примеси
Испытуемый раствор.
Растворяют 30,0 мг
исследуемого вещества в
подвижной фазе и доводят
объем до 100,0 мл тем же
растворителем.
Reference solution B. Dissolve
5.0 mg of ceftriaxone sodium
RS and 5.0 mg of ceftriaxone
impurity A RS in the mobile
phase and dilute to 100.0 ml
with the same solvent.
Стандартный раствор Б.
Раствор сравнения B.
Растворяют 5,0 мг цефтриаксона Растворяют 5,0 мг СО
натрия СО и 5,0 мг цефтриаксона цефтриаксона натрия и 5,0 мг
примеси A СО в подвижной фазе, СО примеси A в подвижной
разбавляют до 100,0 мл тем же
фазе, доводят объем до 100,0 мл
растворителем.
тем же растворителем.
4.
ОригиналПереводчик
Колонка. Pазмеры: 0,25 м × 4,6
мм; неподвижная фаза:
силикагель
октадецилсилильный, размер
частиц 5 мкм.
Mobile phase. Dissolve 2.0 g of Подвижная фаза. Растворяют 2,0 Подвижная фаза. Растворяют
tetradecyl ammonium bromide г тетрадециламмонийбромида в 2,0 г тетрадециламмония
бромида в смеси 440 мл воды, 55
in a mixture of 440 ml of water, смеси 440 мл воды, 55 мл
55 ml of phosphate buffer pH
фосфатного буфера с pH 7,0, 5,0 мл фосфатного буферного
7.0, 5.0 ml of citrate buffer pH
мл цитратного буфера с pH 5,0. раствора с pH 7,0, 5,0 мл
цитратного буферного раствора
5.0.
с pH 5,0.
Column. L = 0.25 m, Ø = 4.6
mm; stationary phase:
octadecylsilyl silica gel (5 µm).
Колонка. Pазмеры: длина 0,25 м,
диаметр 4,6 мм; Неподвижная
фаза: октадецилсиликагель,
размер частиц 5 мкм.
Редактор
5.
ОригиналПереводчик
Detector: UV. Wavelength: 233 Детектор: ультрафиолетовый.
nm. Injection: 20 µl. Flow rate: Длина волны: 233 нм. Объем
1.5 ml/min. Run time: twice the пробы: 20 мкл. Скорость потока:
retention time of ceftriaxone. 1,5 мл/мин. Время
хроматографирования: вдвое
больше, чем время удерживания
цефтриаксона.
System suitability. The
Пригодность
relative retention time should хроматографической системы.
not be less than 1.6 for the
Нескорректированное
ceftriaxone impurity A and for относительное время
ceftriaxone. The resolution
удерживания должно быть не
between these two peaks
менее 1,6 для примеси А
should not be less than 3.
цефтриаксона и цефтриаксона.
Разрешение между этими двумя
пиками должно быть не менее 3.
Редактор
Детектор: УФ. Длина волны
детектирования: 233 нм. Объем
пробы: 20 мкл. Скорость потока:
1,5 мл/мин. Время
хроматографирования:
двукратное время удерживания
цефтриаксона.
Пригодность
хроматографической системы.
Относительное время
удерживания должно
составлять не менее 1,6 для
пиков цефтриаксона и примеси
А. Разрешение между этими
двумя пиками должно быть не
менее 3.
6.
ОригиналПереводчик
Редактор
The column efficiency
should not be less than
2250 theoretical plates,
and the tailing factor for
the ceftriaxone peak is
not more than 2.0.
Inject reference solution
in duplicate, test solution
in triplicate and record
the chromatograms.
Эффективность
хроматографической колонки не
должна быть меньше 2250
теоретических тарелок, а фактор
асимметрии пика цефтриаксона
должен быть не более 2,0.
Определение проводят в двух
параллельных пробах для
раствора сравнения, в трех —
для испытуемого раствора.
Эффективность
хроматографической колонки
должна составлять не менее
2250 теоретических тарелок, а
фактор асимметрии пика
цефтриаксона — не более 2,0.
Раствор сравнения вводят в
двух повторностях,
испытуемый раствор — в трех
повторностях. Снимают
хроматограммы.
7.
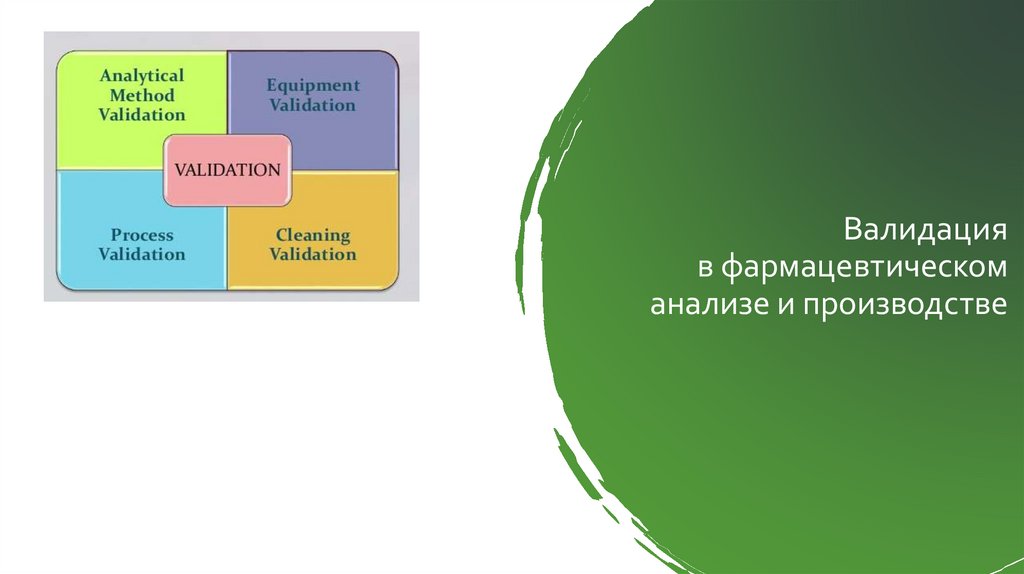
Валидацияв фармацевтическом
анализе и производстве
8.
Валидация аналитических методикЭкспериментальное доказательство того, что методика пригодна для решения предполагаемых задач
Зачем?
для получения надежных и стабильных данных
для определения потенциальных ошибок
для обеспечения качества и безопасности препаратов
по требованию регуляторных органов
Когда?
• перед внедрением в рутинный анализ
• если меняются условия, для которых была проведена валидация
• если меняется методика, и изменение выходит за рамки валидированной сферы применения
ICH Q2 (R1)
9.
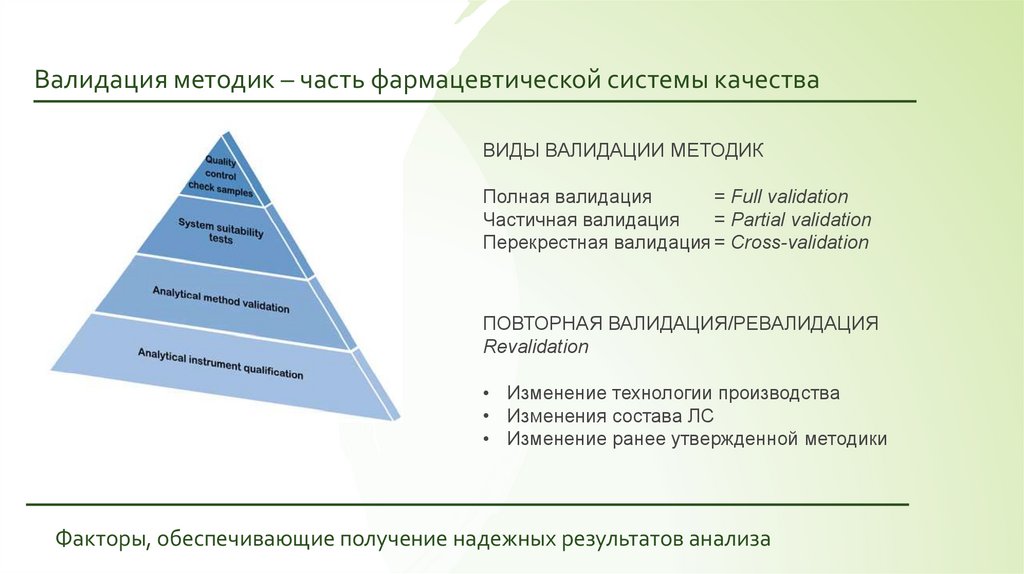
Валидация методик – часть фармацевтической системы качестваВИДЫ ВАЛИДАЦИИ МЕТОДИК
Полная валидация
= Full validation
Частичная валидация
= Partial validation
Перекрестная валидация = Cross-validation
ПОВТОРНАЯ ВАЛИДАЦИЯ/РЕВАЛИДАЦИЯ
Revalidation
• Изменение технологии производства
• Изменения состава ЛС
• Изменение ранее утвержденной методики
Факторы, обеспечивающие получение надежных результатов анализа
10.
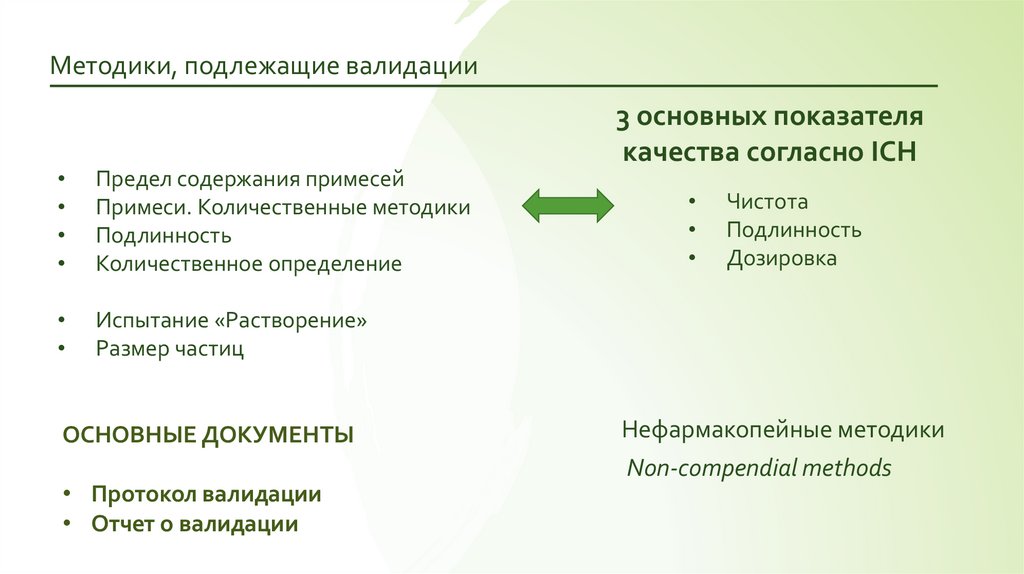
Методики, подлежащие валидации3 основных показателя
качества согласно ICH
Предел содержания примесей
Примеси. Количественные методики
Подлинность
Количественное определение
Испытание «Растворение»
Размер частиц
ОСНОВНЫЕ ДОКУМЕНТЫ
• Протокол валидации
• Отчет о валидации
Чистота
Подлинность
Дозировка
Нефармакопейные методики
Non-compendial methods
11.
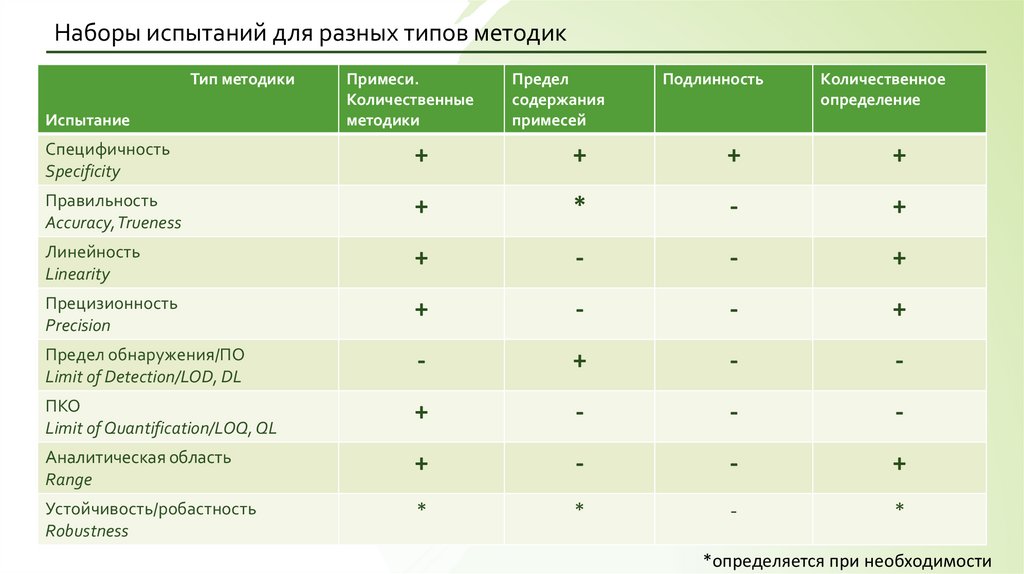
Наборы испытаний для разных типов методикТип методики
Испытание
Примеси.
Количественные
методики
Предел
содержания
примесей
Подлинность
Количественное
определение
Специфичность
Specificity
+
+
+
+
Правильность
Accuracy, Trueness
+
*
-
+
Линейность
Linearity
+
-
-
+
Прецизионность
Precision
+
-
-
+
Предел обнаружения/ПО
Limit of Detection/LOD, DL
-
+
-
-
ПКО
Limit of Quantification/LOQ, QL
+
-
-
-
Аналитическая область
Range
+
-
-
+
Устойчивость/робастность
Robustness
*
*
-
*
*определяется при необходимости
12.
СпецифичностьSpecificity
Способность аналитической методики однозначно оценивать определяемое вещество независимо
от других веществ, присутствующих в испытуемом образце
IDENTIFICATION
подтверждение того, что методика идентифицировать именно определяемое вещество
IMPURITIES
Подтверждение того, что методика позволяет правильно распознать примеси в образце
ASSAY
подтверждение того, что методика позволяет установить содержание или активность именно
определяемого вещества в образце
NB!
Не путать с селективностью
Selectivity
Селективность
Специфичность
13.
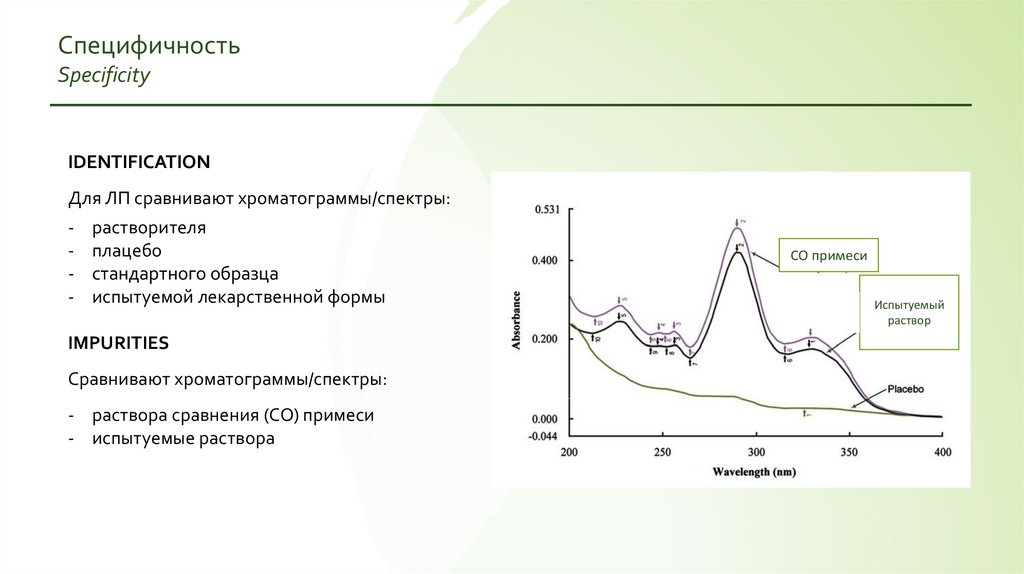
СпецифичностьSpecificity
IDENTIFICATION
Для ЛП сравнивают хроматограммы/спектры:
-
растворителя
плацебо
стандартного образца
испытуемой лекарственной формы
IMPURITIES
Сравнивают хроматограммы/спектры:
- раствора сравнения (СО) примеси
- испытуемые раствора
СО примеси
Испытуемый
раствор
14.
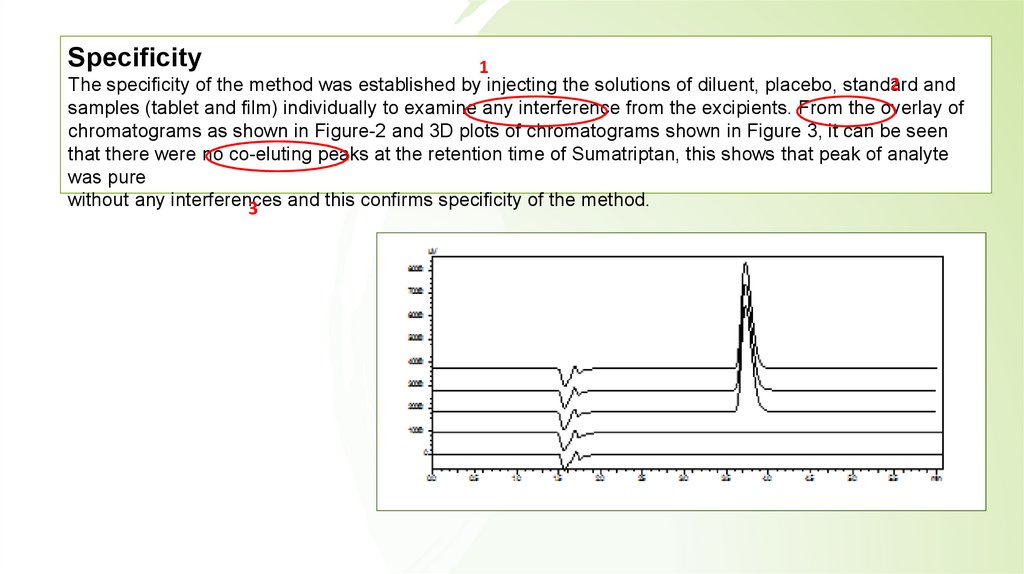
Specificity1
2 and
The specificity of the method was established by injecting the solutions of diluent, placebo, standard
samples (tablet and film) individually to examine any interference from the excipients. From the overlay of
chromatograms as shown in Figure-2 and 3D plots of chromatograms shown in Figure 3, it can be seen
that there were no co-eluting peaks at the retention time of Sumatriptan, this shows that peak of analyte
was pure
without any interferences
and this confirms specificity of the method.
3
15.
ЛинейностьLinearity
Наличие линейной зависимости
аналитического сигнала от концентрации
определяемого вещества в образце
в пределах аналитической области
методики
Характеризует изменение сигнала в
зависимости от концентрации
определяемого вещества
16.
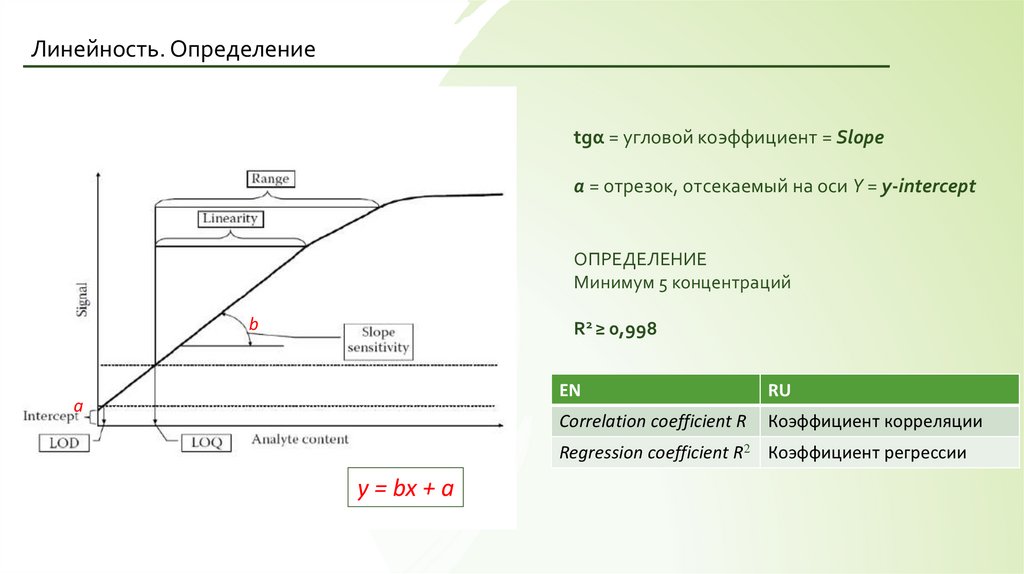
Линейность. Определениеtgα = угловой коэффициент = Slope
a = отрезок, отсекаемый на оси Y = y-intercept
ОПРЕДЕЛЕНИЕ
Минимум 5 концентраций
b
R2 ≥ 0,998
a
EN
RU
Correlation coefficient R
Коэффициент корреляции
Regression coefficient R2 Коэффициент регрессии
y = bx + a
17.
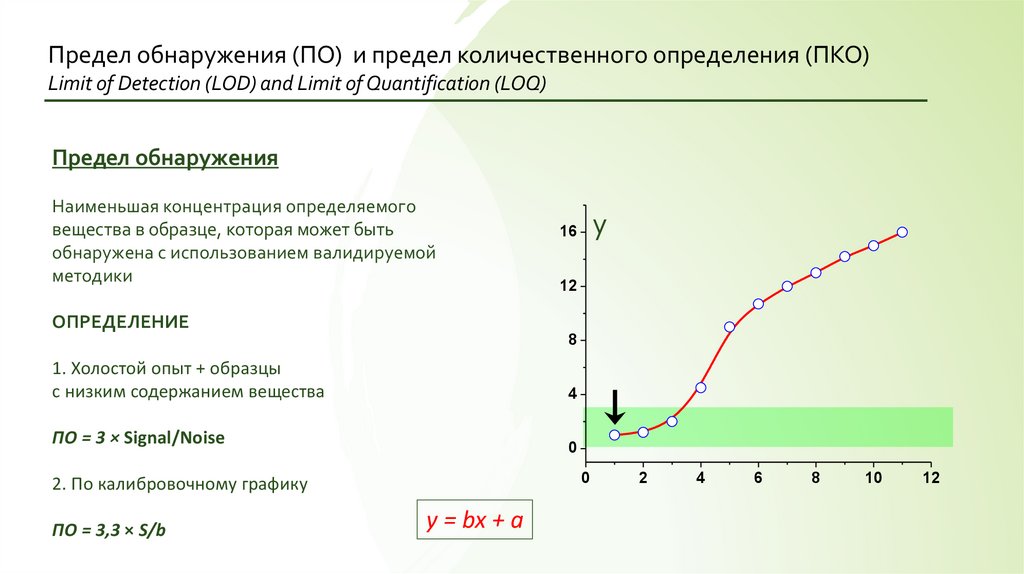
Предел обнаружения (ПО) и предел количественного определения (ПКО)Limit of Detection (LOD) and Limit of Quantification (LOQ)
Предел обнаружения
Наименьшая концентрация определяемого
вещества в образце, которая может быть
обнаружена с использованием валидируемой
методики
y
16
12
ОПРЕДЕЛЕНИЕ
8
1. Холостой опыт + образцы
с низким содержанием вещества
4
ПО = 3 × Signal/Noise
0
0
2. По калибровочному графику
ПО = 3,3 × S/b
y = bx + a
2
4
6
8
10
12
18.
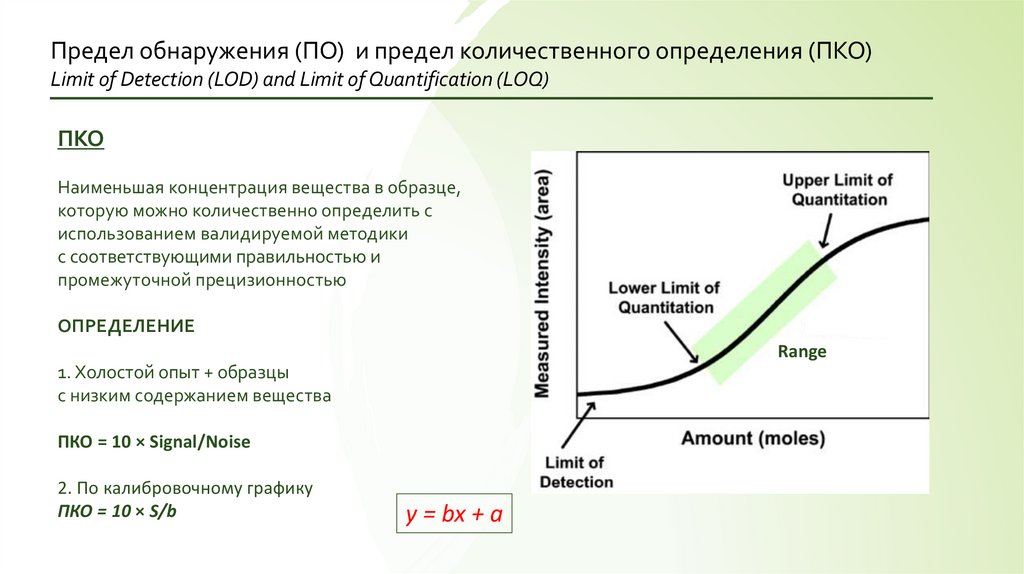
Предел обнаружения (ПО) и предел количественного определения (ПКО)Limit of Detection (LOD) and Limit of Quantification (LOQ)
ПКО
Наименьшая концентрация вещества в образце,
которую можно количественно определить с
использованием валидируемой методики
с соответствующими правильностью и
промежуточной прецизионностью
ОПРЕДЕЛЕНИЕ
Range
1. Холостой опыт + образцы
с низким содержанием вещества
ПКО = 10 × Signal/Noise
2. По калибровочному графику
ПКО = 10 × S/b
y = bx + a
19.
Аналитическая область/диапазон примененияRange
Интервал между наибольшей и
наименьшей концентрациями
определяемого вещества в образце
12
9
Должно быть доказано,
что аналитическая методика
является прецизионной, правильной
и линейной в данном диапазоне
6
3
0
3
6
9
12
20.
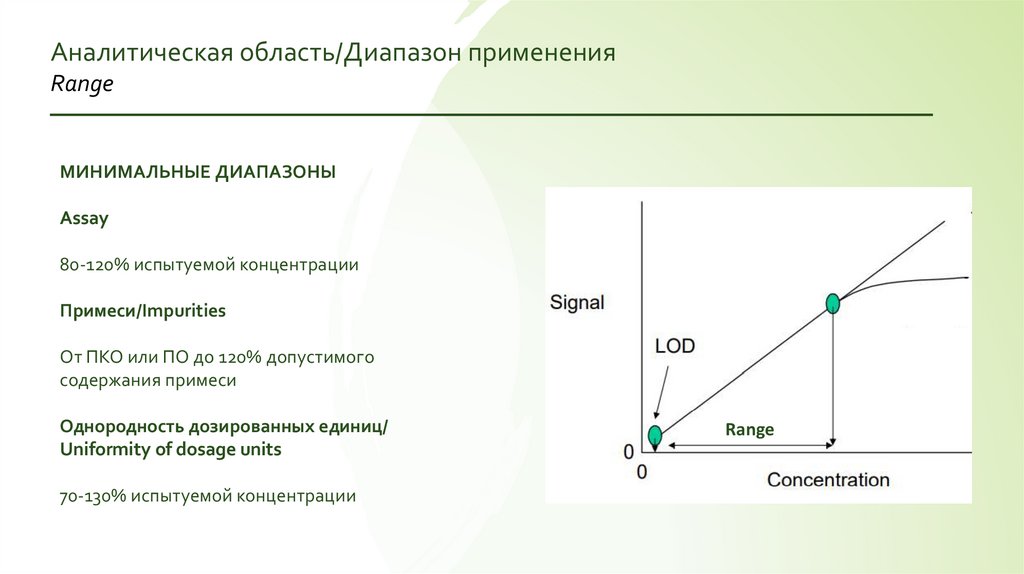
Аналитическая область/Диапазон примененияRange
МИНИМАЛЬНЫЕ ДИАПАЗОНЫ
Assay
80-120% испытуемой концентрации
Примеси/Impurities
От ПКО или ПО до 120% допустимого
содержания примеси
Однородность дозированных единиц/
Uniformity of dosage units
70-130% испытуемой концентрации
Range
21.
ПрецизионностьPrecision
Выражает близость (степень разброса) результатов между сериями измерений, проведенными на
множестве проб, взятых из одной и той же однородной пробы, в предписанных методикой условиях
Три уровня:
1. повторяемость
2. промежуточная прецизионность
3. воспроизводимость
Degree of scatter = степень разброса
22.
1. ПовторяемостьRepeatability, Intra-assay Precision
Прецизионность методики при выполнении повторных испытаний в одинаковых рабочих условиях в
течение короткого промежутка времени
Минимум 3 измерения при, как минимум, 3 концентрациях
или
Минимум 6 измерений при концентрации испытуемого вещества 100%
2. Промежуточная (внутрилабораторная) прецизионность
Intermediate Precision, Inter-assay Precision
Оценивается в условиях работы одной лаборатории
Прецизионность
23.
3. ВоспроизводимостьReproducibility
Прецизионность в межлабораторных испытаниях
Отражает случайные ошибки измерения и показывает степень разброса
параллельных (повторных) измерений, произведенных в разных
лабораториях
Прецизионность
24.
ПрецизионностьPrecision
Прецизионность выражается следующими величинами:
• дисперсия S2
• относительное стандартное отклонение (ОСО)/
коэффициент вариации (КВ)
Sδ,% = ОСО = RSD
CV = КВ
25.
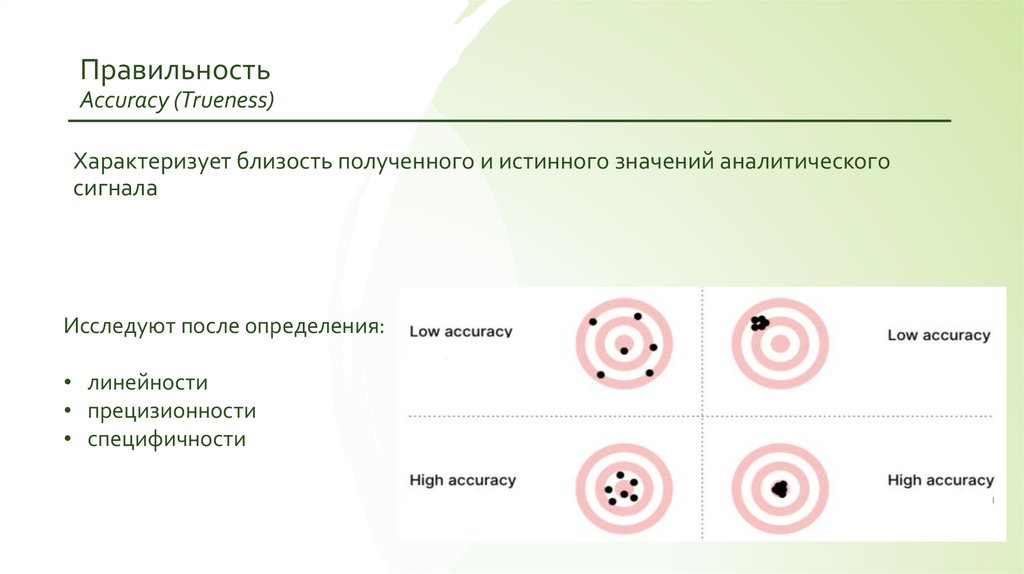
ПравильностьAccuracy (Trueness)
Характеризует близость полученного и истинного значений аналитического
сигнала
Исследуют после определения:
• линейности
• прецизионности
• специфичности
26.
Правильность. Валидация методикОСНОВНОЙ ПОКАЗАТЕЛЬ
Открываемость (Recovery)
Соотношение между полученным
средним и истинным/опорным
значениями с учетом
соответствующих
доверительных интервалов
27.
Правильность. Валидация методикПРОЦЕДУРА
не менее 9 определений (по 3 на каждом
уровне концентраций)
Диапазон концентраций должен покрывать
валидируемый диапазон
ИЗМЕРЕНИЕ
Открываемость, % содержания точно
известной концентрации примеси,
добавленной к образцу (98-102%)
Accuracy
Accuracy of the method was examined by spiking the
known quantities of standard at 80%, 100%, 120% to
the drug product solution and were analyzed in
triplicate. The % RSD and the % recovery were within
the acceptable limit indicating that the proposed
method is accurate.
28.
Правильность методикиAccuracy
This study was carried out using pre-formulated granules containing pure levofloxacin hemihydrate and
common excipients. Calculation was done from the label claim and the average weight of the final product.
Previously used dilution pattern was followed for the granules to obtain five concentrations — 80%, 90%,
100%, 110% and 120% of reference solution.
29.
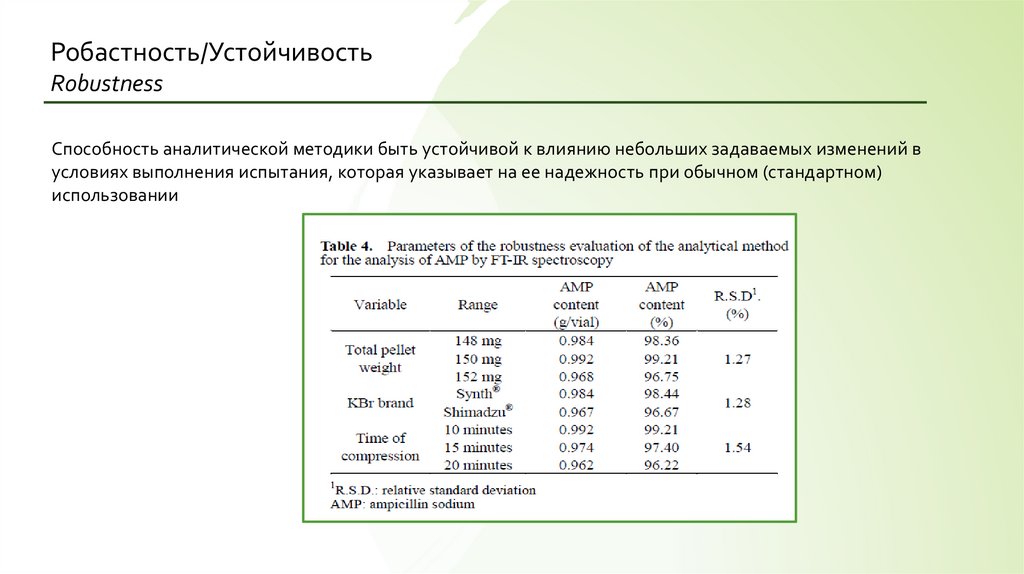
Робастность/УстойчивостьRobustness
Способность аналитической методики быть устойчивой к влиянию небольших задаваемых изменений в
условиях выполнения испытания, которая указывает на ее надежность при обычном (стандартном)
использовании
30.
Базовые понятиястатистики в валидации
31.
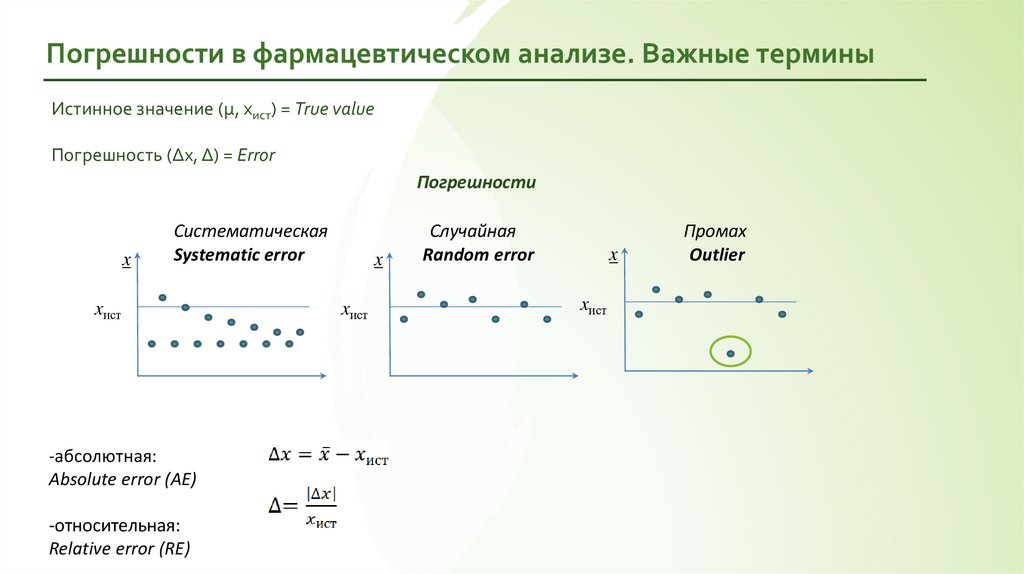
Погрешности в фармацевтическом анализе. Важные терминыИстинное значение (μ, xист) = True value
Погрешность (Δx, Δ) = Error
Погрешности
x
Систематическая
Systematic error
xист
-абсолютная:
Absolute error (AE)
-относительная:
Relative error (RE)
x
xист
Случайная
Random error
x
xист
Промах
Outlier
32.
Погрешности в фармацевтическом анализеEN
Out-of-Specification (OOS) Result
RU
Результат, выходящий за пределы спецификации
Результат, выходящий за ожидаемые пределы
(но не выходящий за пределы спецификации)
Out of Trend (OOT) Result
Результат, выходящий за пределы тенденций
(трендов)
Причины:
-
ошибка лабораторного анализа
ошибка отбора проб
несоответствие продукта требованиям
33.
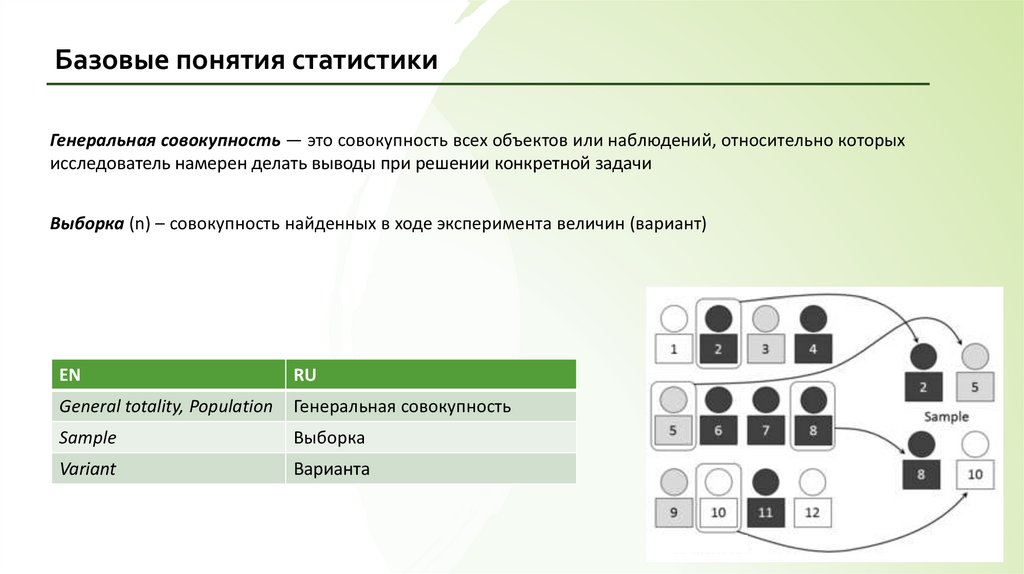
Базовые понятия статистикиГенеральная совокупность — это совокупность всех объектов или наблюдений, относительно которых
исследователь намерен делать выводы при решении конкретной задачи
Выборка (n) – совокупность найденных в ходе эксперимента величин (вариант)
EN
RU
General totality, Population
Генеральная совокупность
Sample
Выборка
Variant
Варианта
34.
Базовые понятия статистикиСтандартное отклонение – характеризует разброс вокруг среднего
Дисперсия – мера воспроизводимости результатов в выборке
Относительное стандартное отклонение (коэффициент вариации) – показатель рассеивания
значений результатов относительно математического ожидания, мера неопределенности
Среднее:
, n – число
измерений
35.
Базовые понятия статистики• Среднее выборки
– приближенное значение
• Достоверность оценки – величина доверительного интервала
• То есть истинное значение находится в интервале
EN
RU
Confidence level
Доверительная вероятность
Confidence interval
Доверительный интервал
Перевод:
- доверительный интервал (ДИ) 95%
- 95% доверительный интервал (ДИ)
«Мы на 95% уверены, что содержание АФИ
в готовом лекарственном препарате составляет
от 95,0% до 105,5%
«Параметр находится где-то здесь
с вероятностью 95% »
95,0%
105,0%
36.
Q-критерий/контрольный критерий идентификации грубых ошибокDixon's Q test
Метод определения выбросов/промахов (Outliers)
Если Qрасч > Q крит– результат отбрасывается
Где
Х? - "подозрительное" значение (вероятный промах)
Хближайшее - ближайшее к подозрительному значение
Хmin и Хmax - максимальное и минимальное значения ряда значений
n = 3..7
37.
Контакты преподавателя:Анна Трифонова
+ 375 44 729 46 84
a.trifonova@pharmconsult.org
www.pharmconsult.org
СПАСИБО ЗА
ВНИМАНИЕ!