






 факторы гемобластозов")

")
")




















")

")




")

















































- «селезеночная лейкемия» - первый описанный гемобластоз человека. Частота ХМЛ: 1–1,5 слу")



















 специфически ингибирует протеинкиназу, продуцируемую химерным геном BCR-ABL, что приводит к блокаде пролиферации и")


 (болезнь Вакеза, polycythemia vera)")







")

")







 Медицина
МедицинаПохожие презентации:
")

")







Острые лейкозы
1.
Лекциядля 5 курса 4 факультета ФПВ на тему:
« Острые лейкозы »
Лектор- доцент кафедры ВМТ
Шарова Н.В.
2.
Онкогематологическиезаболевания
(гемобластозы) – злокачественные новообразования,
морфологическим
субстратом
которых
являются
неопластически трансформированные кроветворные
клетки
.
Гемобластозы, при которых костный мозг первично
повсеместно заселен опухолевыми клетками, называют
лейкозами
Термин “лейкемия” принадлежит R.Virchow (в 1845
описал ХМЛ)
3. Основные формы гемобластозов по классификации опухолей ВОЗ 2000 года
• - острые миелоидные лейкозы• - хронические миелопролиферативные заболевания
(хронический миелолейкоз, истинная полицитемия,
сублейкемический миелоз, эссециальная
тромбоцитемия)
- В-клеточные опухоли (острый В-клеточный
- лимфобластный лейкоз, хронический лимфолейкоз,
множественная миелома и др.
- Опухоли из Т- и NK-клеток (острый Т-клеточный
лимфобластный лейкоз и др.)
- Лимфома Ходжкина (лимфогранулематоз)
- Неходжкинские лимфомы (НХЛ)
- миелодиспластические синдромы
4.
• Максимов А.А. в 1908гвпервые высказал
представление о наличии
единой клеткипредшественницы для всех
ростков кроветворения –
впоследствии СКК –
стволовая клетка крови.
5.
6. Клеточный состав костного мозга %
• Клеточный состав костного мозга – 50 – 250 кл в 1 мкл• Мегакариоциты - 0,2-0,6
• Бласты – 0,1 -1,6%
Миелобласты – 0,2-1,7
Лимфоциты- 4,3-13,7
Промиелоциты – 1,0-4,1
Моноциты – 0,7-3,1
Миелоциты – 7,0 – 12,2
Метамиелоциты – 8,0- 15,0
Палочкоядерные – 12,8-23,7
Сегментоядерные 13,1-24,1
Всего нейтрофильного ряда- 52,7-68,9
Эозинофилы – 0,5-5,8
Базофилы – 0-0,5
Эритробласты- 0,2-1,1
Пронормоциты- 0,1-1,2
Нормоциты базофильные -1,4-4,6
Нормоциты полихроматофильные 8,9-16,9
Нормоциты оксифильные - 0,8-5,6
Всего клеток эритроидного ряда – 14,5 – 26,5
• ЛЭС – 2,1 – 4,5
7. Распространенность гемобластозов
• Суммарная частота гемобластозов: 25–30 случаев на100
000 населения в год
• Наиболее распространены неходжкинские лимфомы (10–12
случаев). В последние 20–30 лет частота неходжкинских
лимфом постоянно увеличивается, заболеваемость другими
гемобластозами находится на стабильном уровне.
8. Распределение гемобластозов
• Все онкогематологические заболевания, за исключением остроголимфобластного лейкоза, лимфобластной лимфомы и лимфомы
Беркитта, значительно чаще возникают
у взрослых
• Некоторые нозологические формы (хронический лимфолейкоз,
множественная миелома, истинная полицитемия, сублейкемический
миелоз) развиваются только в пожилом возрасте.
• Острый
лейкоз
заболевание у детей.
основное
злокачественное
• Большинство гемобластозов, особенно лимфопролиферативные
заболевания, достоверно чаще возникают у европеоидов (прежде
всего у семитов – арабов и евреев), существенно реже – у негроидов и
крайне редко – у лиц азиатского происхождения
9. Этиологические (предрасполагающие) факторы гемобластозов
• В основе -первичная мутации кроветворной клеткипредшественницы под действием мутагенов – хромосомные
поломки.
Ионизирующая радиация.
Химические соединения.
Генетические заболевания (7% всех опухолевых больных, в семьях с
ОЛ заболеваемость выше в 3-4 раза).
Связь с аутоиммунными заболеваниями , наследственными и
приобретенными иммунодифицитами.
Вирусы.
Длительное воздействие бактериальных Аг
+ Возрастное истощение противоопухолевого иммунитета (гена 53,
ответственного за апоптоз).
предшествующие
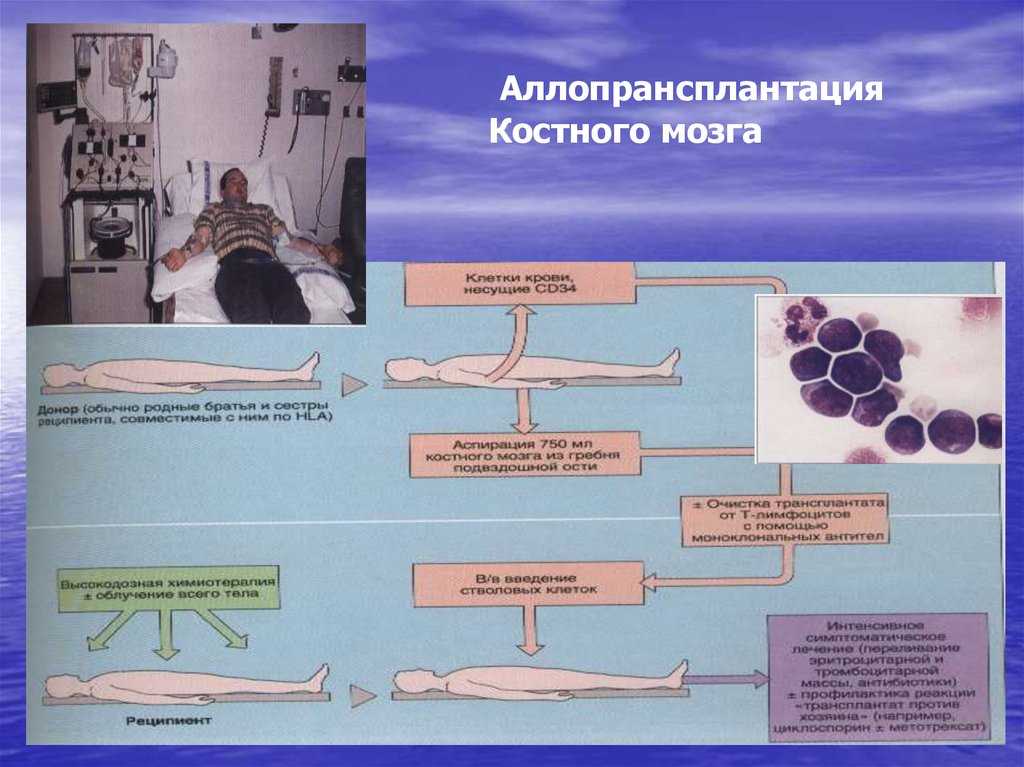
заболевания
системы
кроветворения
(рефрактерные формы анемий, пароксизмальная ночная гемоглобинурия, так
называемые миелодисплазии).
10. Патогенез онкогематологических заболеваний
Гемобластозы возникают вследствие мутации
клетки-предшественницы, приводящей к клональной
неопластической пролиферации злокачественно
трансформированных кроветворных клеток на любом
уровне их дифференцировки (полипотентность)!!,
характеризующихся:
1) блокадой дифференцировки;
2) способностью к неограниченному делению –
увеличенное количество митозов
. 3) большей продолжительностью жизни, поэтому они
быстро накапливаются в организме
11. Патогенез онкогематологических заболеваний (продолжение)
• При лабораторных признаках лейкозов в организме больногоимеется около 1011 бластных клеток, при развитии клинических
симптомов – 1012 бластов (1кг).
• При злокачественных лимфомах клинические симптомы
(прежде всего – лимфоаденопатия) появляются при значительно
меньшем объеме опухолевой массы
• Развитие большинства клинико-лабораторных проявлений
(анемия, геморрагический синдром, лихорадка, увеличение
размеров лимфатических узлов, селезенки) обусловлено
“вытеснением”
нормальной
гемопоэтической
ткани
опухолевыми клетками или выделением факторов, тормозящих
эритро- и тромбоцитопоэз.
12. Патогенез онкогематологических заболеваний (окончание)
• Для гемобластозов характерна опухолеваяпрогрессия:
по мере развития заболевания появляются клоны более
злокачественных клеток с новыми свойствами (морфологическими,
цитохимическими, иммунологическими и т.д.), что объясняет развитие
резистентности к ранее эффективному лечению.
Злокачественные клетки активно метастазируют за пределы
органов кроветворения, что приводит к развитию опухолевой
инфильтрации внутренних органов, лимфатической ткани, кожи,
слизистых оболочек, оболочек и вещества головного и спинного мозга
и может сопровождаться органомегалией, функциональной
недостаточностью внутренних органов, лимфоаденопатией,
гиперпластическим гингивитом, лейкемидами кожи, нейролейкозом и
т.д.
13.
Острые лейкозы – группа клональныхзаболеваний системы крови, первично
поражающих костный мозг, морфологическим
субстратом которых являются злокачественно
трансформированные бластные
клетки, неспособные к нормальной
дифференцировке до зрелых клеточных форм, с
вытеснением ими нормальных элементов и
инфильтрацией ими различных органов и тканей.
14.
• Все острые лейкозы возникают из одной мутировавшей клетки,которая может относиться как к очень ранним, так и
коммитированным в направлении различных линий
кроветворения.
• Клиническое течение острого лейкоза,
терапия, эффективность лечения и прогноз
заболевания определяются:
• 1) принадлежностью бластных клеток к той или иной линии
кроветворения,
• 2) степенью их дифференцировки ,
• 3) степенью вытеснения нормальных ростков кроветворения
обуславливают
15.
Острые
лейкозы
–
отдельная
группа
онкогематологических заболеваний, которые никогда не
трансформируются в хронические лейкозы.
Острый
лейкоз
морфологический.
–
прежде
всего
диагноз
• Острый лейкоз может быть первичным и вторичным
• ( после цитостатической или лучевой терапии).
• Острый лейкоз составляет 3% всех злокачественных
опухолей человека.
• Частота острых лейкозов в среднем составляет 7,7-13,0
случаев на 100 000 населения в год и приблизительно
одинакова в различных регионах.
Соотношение М:Ж при ОЛ = 13,2 : 7,7
16.
Выделяют два вида заболевания, которые различаютсяпо
морфологии,
течению,
характеру
проводимой
химиотерапии и результатам лечения:
1)
Острый
лимфобластный
лейкоз
(80–90% в структуре лейкозов детей)
• 2) острые (нелимфобластные, миелобластные) лейкозы
• (в 80% случаев развиваются у взрослых)
• Среднее соотношение ОМЛ : ОЛЛ = 6:1
17. FAB - классификация острых лейкозов
В 1976 году группой экспертов Франции, США и Великобритании(FAB) была предложена классификация ОЛ, которая основана на
1) морфологических ,
2) цитохимических особенностях,
3) иммунофенотипировании бластных клеток
В соответствии с FAB-классификацией
8 типов ОМЛ (М0–М7) и 3 типа ОЛЛ (L1–L3).
Классификация ВОЗ 1991г учитывает цитогенетические и
молекулярно-генетические показатели, основана на меньшем
количестве клеток, позволяет выделить ряд подтипов,
биологических подгрупп, тем самым индивидуализировать
лечение больных.
18.
• Для верификации вариантаострого лейкоза и его
варианта требуются
цитохимические,
результаты которых имеют
прогностическое значение и
позволяют планировать
лечебную тактику.
Положительная шик-реакция на гликоген- положительная при ОЛЛ
+реакция на кислую фосфатазу
19. Иммунофенотипирование
Внедрение в практику моноклональных антител
позволило идентифицировать специфические рецепторы,
антигены и другие молекулы (маркеры) на мембране
и/или в цитоплазме бластных клеток.
В настоящее время идентифицировано более 200 маркеров,
получивших название кластеров дифференцировки (CD).
Иммунофенотипирование необходимо для
1) подтверждение диагноза
2) установление диагноза при сомнительных морфологических и
цитохимических результатах
3) мониторинг для выделения новых более злокачественных клеток;
4) разделение ОЛ на различные прогностические группы
• Пример: острый недифференцированный лейкоз - HLA-DR+,
CD15+|-, CD13+\-, CD33+\-
20. Иммунофенотипическая характеристика ОМЛ
Иммуноподвариант лейкозаКлеточный фенотип
М0- малодифференцированный
лейкоз
HLA-DR+, CD15+|-,
CD13+\-, CD33+\-
М1-миелобластный лейкоз без
созревания
HLA-DR+\-, CD38+|-,
CD11a+\-, RFB1+\- CD53
М2-миелобластный лейкоз с
созреванием
HLA-DR+\-CD72+,CD38+\,CD53+
М3-промиелоцитарный лейкоз
CD53+,RFB1+\-,CD11b+\-
М4- миеломонобластный
HLA-DR+,CD15+,CD38-,
М5-монобластный
HLA-DR+,CD11b+,CD15
М6-эритромиелоз
HLA=DR+\-,CD38-,
М7-мегакариобластный
CD38+,CD41+,HLA-DR+\-
частота
21. Иммунологические и цитогенетические варианты острых лимфобластных лейкозов
Тип и частотаИммунофенотип
Цитогенетические
нарушения
Пре-преВ-ОЛЛ (ранний HLA-DR,TdT+, CD34+,
пре-преВ 5-10% и
CD10+\-,CD19+,cIg-,sIgCommon 45%
,CD20-\+,CD24+\-
T(12,21)d 2025%,t(9,22),11g23
Пре-В-ОЛЛ – 20%
HLA-DR, CD19,CD20+\,CD24,CD9,CD10,CD34-
T(1,19)
В-ОЛЛ – 4-5%
HLADR,CD19,CD20,CD22,CD24
T(8,14),t(2,8),t(8,22)
Т-ОЛЛ -20-31%
HLA-DR+\,CD1,CD2,CD3,CD5,CD7,
T(1,14)d 15-25%
22. Цитогенетическое исследование
• У 90% больных ОЛ находят• генетические поломки
(транслокации, делеции,
инверсии, гиперплоидию…).
При ОЛЛ транслокация (9;22)
или
(4;11) – фактор неблагоприятного
прогноза. Гиперплоидия
характерна
для благоприятного течения ОЛ
(часто у детей с ОЛЛ).
23. Основные клинические синдромы при ОЛ
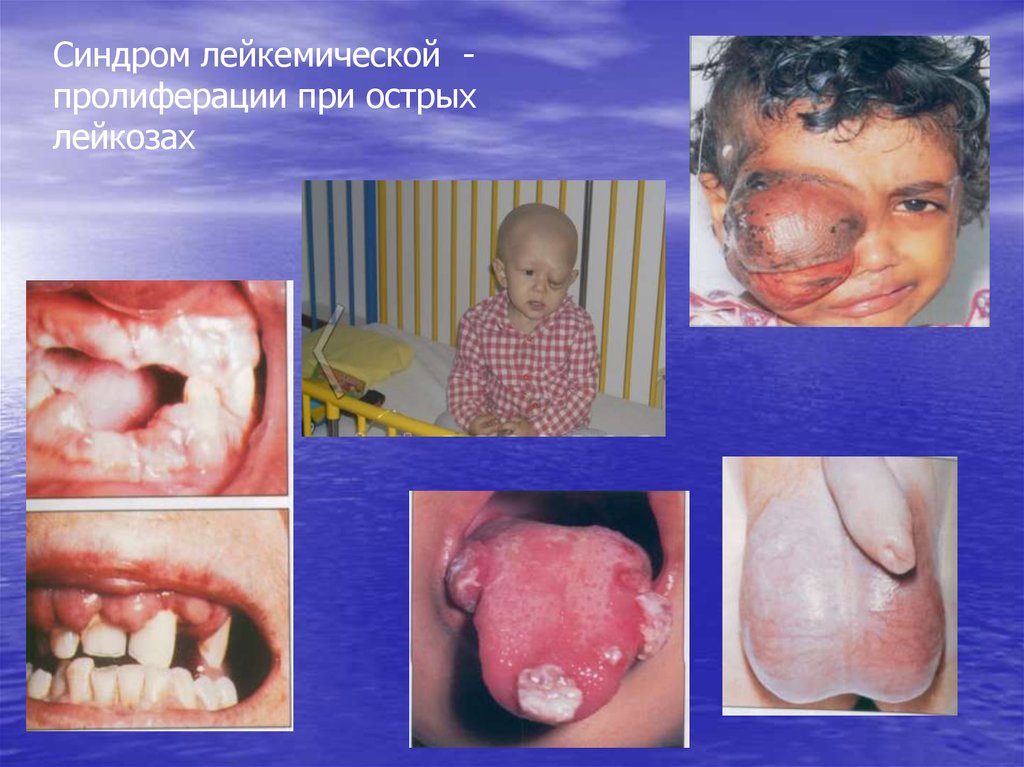
• Синдром лейкемической пролиферации: лимфоаденопатия,
гепатоспленомегалия, расширение средостения; инфильтрация десен и
яичек, лейкемиды кожи; поражение мозговых оболочек и черепномозговых нервов (нейролейкоз – чаще при ОЛЛ)
• Синдром опухолевой интоксикации (снижение массы тела,
общая слабость, усиленная потливость, повышение Т тела, болезненность
костей)
• Анемический синдром (бледность, тахикардия, одышка)
• Геморрагический синдром (экхимозы, петехии,
• внутренние кровотечения), ДВС-синдром
• Синдром инфекционных осложнений (стоматит,
некротизирующая ангина пневмония, сепсис, абсцессы)
24.
Синдром лейкемической пролиферации при острыхлейкозах
25.
Проявления геморрагическогоcиндрома при ОЛ, в том числе
в головной мозг, ЖК-тракт
Инфекционные осложнения
При ОЛ
26. Нейролейкоз Инфильтрация мозговых оболочек, приведшая к поражению лицевого нерва и одностороннему параличу мимических мышц
Отек и кровоизлияния всетчатке в результате
диффузной
инфильтрации мозговых
оболочек
27. Варианты начала острого лейкоза
• - примерно у 50% больных начинается остро с клиники ангины,пневмонии, инфекции мочевыделительных путей;
• - у 25-30% больных постепенное начало, проявляющееся слабостью,
повышенной утомляемостью, нарастанием
• кардиальных проявлений (анемическим синдромом),
болями в суставах и мышцах;
ноющими
• - в 10% случаях заболевание манифестирует
геморрагическими проявлениями;
• - увеличение периферических лимфоузлов, селезенки,
печени;
• - реже дебютом могут быть стоматит, мигрирующий тромбофлебит,
специфические кожные проявления, внезапные параличи или парезы.
• - 5% (до 52 %) - выявляется при случайном исследовании крови.
28. Особенности течения некоторых форм ОМЛ
• О недифференцированный лейкоз (М0): частота его уменьшается всвязи появляющимися дополнительными маркерами ОЛ при
иммунофенотипировании.
• О. миелобластный лейкоз (М1+М2)- 50-60% всех случаев ОЛ.
Заболевание чаще встречается у лиц старше 40 лет;
характеризуется быстрым развитием всех синдромов:
интоксикационного, анемического, геморрагического синдрома.
• О. Промиелоцитарный лейкоз (М3)- встречается у 5% больных.
Характерно развитие тяжелого ГЕМОРРАГИЧЕСКОГО СИНДРОМА, ДВСсиндрома, при адекватной терапии которого прогноз б-го = ОЛЛ
• О. миеломонобластный лейкоз (М4) -15%, течение более
агрессивное, чем М1. Нередко развиваются экстрамедуллярные
очаги (нейролейкз, лейкемиды кожи, гипертрофический гингивит).
29.
• О. монобластный лейкоз (М5) – 10%, клиника схожа с М4,экстрамедуллярные очаги встречаются еще чаще- до 30%.
• .
• О. Эритромиелоз (М6)- 5%. –острый эритробластый лейкоз –
болезнь Ди Гульельмо. Начальным проявлением может быть
анемия. В костном мозге картина напоминает В12-дефицитную
анемию. Помогает диагностике увеличение % бластных клеток.
Нередко встречаются «ревматические» симптомы: серозиты,
артралгии; лечение менее эффективно.
• О. Мегакариоцитарный лейкоз (М7) – наиболее редкая форма
острого лейкоза, характерно развитие миелофиброза,
затрудняющее получение костного мозга. Нередко развивается
гипертромбоцитоз.
30.
• О. Лимфобластный лейкоз – 10-15% (до 25%)взрослых больных ОЛ, (85% детей).
• Характено частое развитие синдрома лейкемической
пролиферации и нейролейкоза.
• Интоксикационный, анемический, геморрагический и синдром
инфекционных осложнений встречаются значительно реже.
• Более чем у 50% больных встречаются изменения кариотипа:
при этом гиперплоидия сопровождается хорошим ответом на
терапию.
• Особенно неблагоприятно течение при наличии
филадельфийской хромосомы .
• Лечение ОЛЛ проводится по другим программам химиотерапии,
поэтому д/д между ними строго обязателен.
31. Клинический анализ крови
• Количество лейкоцитов периферической крови чащеостается на сублейкемическом уровне и не превышает 20–
30х109/л (может варьировать в очень широких пределах (от
1х109/л до 200х109/л)
• - тромбоцитопения (менее 50х109/л) - у 25-50% больных
• - нормохромная нормоцитарная анемия на момент
диагностики заболевания имеется у большинства больных
32. Пример клинического анализа крови больного ОЛ
Гемоглобин – 79г/л;
эритроциты – 2,8×10.12;
цветовой показатель- 0,9;
лейкоциты – 10.0 ×10.9;
Базофилы – 0%;
эозинофилы – 0%;
Бласты – 37%;
Промиелоциты – 0%
Миелоциты – 0%
Метамиелоциты – 0%
нейтрофилы палочкоядерные -1%;
сегментоядерные – 12%; (1300),
лимфоциты -45%;
моноциты – 5%.
Тромбоциты – 30.000
“лейкемический
провал” hiatus leukaemicus
33.
• В типичных случаях между бластами и зрелымигранулоцитами отсутствуют промежуточные формы
клеток нейтрофильного ряда (“лейкемический провал”
или hiatus leukaemicus).
• При уровне лейкоцитов свыше 100х109/л резко
повышается риск развития лейкостатических
осложнений (неврологические нарушения, острый
респираторный дистресс-синдром, у мужчин –
приапизм).
• Биохимическое исследование: повышение уровня
ЛДГ, мочевой кислоты.
34. Клинический анализ крови- ( морфологическое исследование)
Клинический анализ крови( морфологическое исследование)• При подсчете лейкоцитарной
формулы у 90% больных
выявляются бластные
клетки,
число
которых может колебаться от
1–2 до 100%.
• Диагностически значимо – 30%.
• Принадлежность клеток к
миелоидной или лимфоидной
линии МОРФОЛОГИЧЕСКИ
возможна в 70%
35. Проведение костномозговой пункции обязательно!!!
• Иглы для проведения пункции:Игла Кассирского
Современные иглы
36. Техника проведения стернальной пункции (трепанобиопсии)
Область грудиныОбласть подвздошной кости
37. Миелограмма- обязательна !
• Количествомиелокариоцитов
обычно повышено,
мегакариоциты
отсутствуют или
их количество
снижено,
сужение
нормальных
ростков гемопоэза,
Бласты – чаще всего
более 20%- 30% (клетки
с ядрышками в центре)
38. Для проведения морфологического исследования используются:
• 1 Клетки периферической крови• 2 Мазки-отпечатки лимфоузла (опухоли) на предметном стекле
• 3 Пункционная аспирационная биопсия лимфоузла или
тотальная биопсия
• 4 Цитологическое исследование костного мозга: миелограмма,
трепанобиопсия с иммунофенотипированием клеток,
• 5 Цитологическое исследование биожидкостей (спинномозговой ликвор, плевральная жидкость)
39.
• Существует правило:необходимо в течение всей жизни больного ОЛ хранить
пунктаты костного мозга и мазки периферической крови, так
как под влиянием проводимой цитостатической терапии картина
крови и костного мозга становится нетипичной для ОЛ (в
препаратах появляются так называемые терапевтические
бласты с более грубым ядром и их практически нельзя отличить
от лимфоцитов).
• При повторных обращениях больного в другие лечебные
учреждения диагноз ОЛ может быть подтвержден лишь при
изучении первичных препаратов.
40. Стадии острого лейкоза
• I.Первая атака (первый острый период) – время от первых
проявлений заболевания до определения результатов индукционной
химиотерапии.
• II. Полная ремиссия – ≥ 4-х недель отсутствуют клинические и
гематологические признаки ОЛ:
а) без очагов экстрамедуллярного лейкемического роста;
б) в периферической крови отсутствуют бластные клетки,
количество нейтрофилов > 1,5х109/л, тромбоцитов > 100х109/л;
в) имеется достаточная клеточность костного мозга (>20% по
данным трепанобиопсии);
г) в миелограмме бластов <5%, представлены все ростки
кроветворения с нормальным созреванием.
41. Стадии острого лейкоза (окончание)
• III. Рецидив – появление клинических и гематологическихпризнаков ОЛ после полной ремиссии различной длительности.
- снижение количества нормальных клеток или появление
бластов в периферической крови;
- изолированный экстрамедуллярный рецидив (нейролейкоз,
лейкозная инфильтрация яичка и др.). При бластозе костного
мозга >5% диагностируется костномозговой рецидив.
Выделяют ранний (длительность полной ремиссии <12
месяцев) и поздний (продолжительность полной ремиссии >18
месяцев) рецидив.
• IV. Терминальная стадия – полная резистентность к
проводимой терапии с ближайшим неблагоприятным
исходом.
42. Дифференциальный диагноз ОЛ
1 - заболевания, не являющимися гемобластозами:
инфекционный мононуклеоз,
Апластическая анемия,
В12-дефицитная анемия.
2 - Гемобластозы:
Хронический лейкоз в стадии бластного криза,
Миелодиспластический синдром
Неходжкинские лимфомы в ст. лейкемизации
Лейкемоидные реакции
Солидные опухоли через 2-3 года после ЛТ или ХТ
• 3. Метастазы в костный мозг
43.
• Прогностические факторы при острыхмиелоидных лейкозах:
• 1) Значимые: - возраст более 60 лет – неблагоприятный
прогноз;
• - хромосомные нарушения: т(8,12) – высокая частота полных
ремиссий, длительная выживаемость;
• - вторичные лейкозы – прогноз неблагоприятный.
• 2) Менее значимые: другие FAB – варианты лейкоза (М4-М7) –
неблагоприятный прогноз.;
• - общесоматический статус (более 50% - благоприятный
прогноз)
• - лейкоциты более 50 в микролитре – благоприятный;
• - наличие палочек Ауэра – благоприятный прогноз.
44.
• Прогностически неблагоприятныефакторы при остром лимфобластном
лейкозе:
-
возраст более 35 лет;
лейкоцитоз более 300 в 1 микролитре;
достижение ремиссии более чем за 4 недели;
хромосомные аномалии (т9;22) и т (4;11);
В-иммунологический вариант лейкоза.
45. Лечение острого лейкоза
• 1 исторический этап – 19 – начало 20 века – определениенозологической формы и отсутствие какого-либо лечения.
Длительность жизни больных – около 3 месяцев!!
• 2 этап – с начала 40-х годов 20 века начали использовать
гемотрансфузии, достигали отдельных клинических ремиссий.
• 3 этап – 1948 год, первое клиническое применение
аминоптерина – цитостатика, антагониста фолиевой кислоты
– привело к первой полной клинико-гематологической
ремиссии.
• В настоящее время – программная (протокольная) система
использования химиотерапевтических препаратов.
• Заслуга во внедрении программной терапии ОЛ принадлежит
академику АМН Воробьеву АИ
46. Принципы химиотерапии острых лейкозов
• 1.Лечение необходимо начинать сразу же после
установления диагноза, ( не только морфологического, но
по результатам иммунофенотипирования, цитогенетики,
молекулярно-биологического исследования) поскольку при
отсутствии терапии продолжительность жизни пациентов
не превышает трех месяцев.
2. Терапия должна проводиться в специализированном
гематологическом
стационаре,
оснащенном
асептическими палатами. Лечение больных острым
лейкозом в терапевтическом отделении недопустимо!!!
47.
• 3.Выбор схемы лечения определяется морфологическим,
цитохимическим и иммунофенотипическим, цитогенетическим
вариантом острого лейкоза.
• 4. В большинстве случаев используется ПХТ: так как
цитостатики действуют на определенные стадии клеточного
митотического цикла, изменение протокола проведения схемы
недопустимо!
48. Основные методы лечения ОЛ
• 1 Цитостатическая терапия– уничтожение лейкозного клона;
- восстановление нормального кроветворения
• 2 Трансплантация костного мозга
• 3 Сопроводительная терапия ( для уменьшения риска
осложнений)
• 4 Лейкоферез
49.
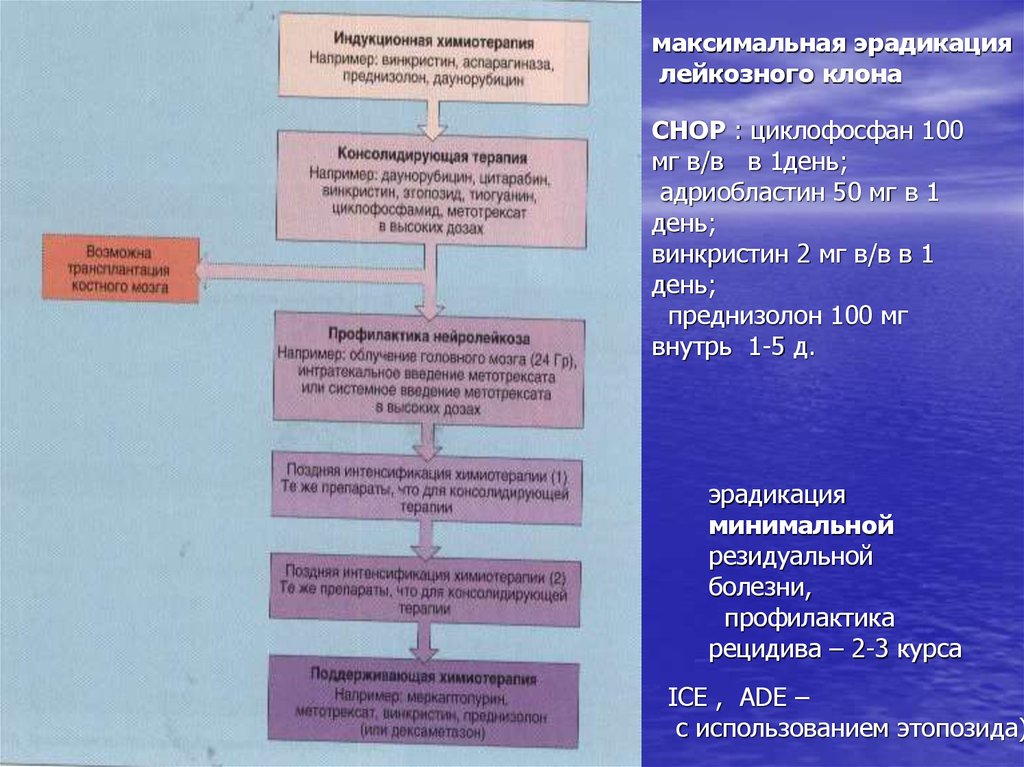
максимальная эрадикацилейкозного клона
ДАТ:
-цитозар 100мг/м2 в/в
2р (1-7 дни)
- даунорубицин 45
мг/м2 в/в 1-3 дни
-тиогуанин 100 мг 2р
(1-7 дни)
эрадикация
минимальной
резидуальной
болезни,
профилактика
рецидива – 2-3 курса
50.
максимальная эрадикациялейкозного клона
CHOP : циклофосфан 100
мг в/в в 1день;
адриобластин 50 мг в 1
день;
винкристин 2 мг в/в в 1
день;
преднизолон 100 мг
внутрь 1-5 д.
эрадикация
минимальной
резидуальной
болезни,
профилактика
рецидива – 2-3 курса
ICE , ADE –
с использованием этопозида)
51. Сопроводительная терапия лейкозов:
• 1 – Дезинтоксикационная терапия (физиологический раствор,полиглюкин, форсированный диурез и др.)
• 2 – Гемокомпонентная терапия ( переливание тромбоцитарной
массы, эритроцитарной массы, очень редко лейкоцитов)
• 3 – Коррекция метаболических нарушений
• ( плазмоферез, гемосорбция и др.)
• 4 – Лечение инфекционных осложнений (антибактериальные,
противогрибковые, противовирусные средства)
• 5 – Аллопуринол для лечения антилейкоцитозного синдрома.
• Антиэметическая терапия
52. Осложнения острых лейкозов
• 1. Инфекционные осложнения (у 70%больных определяют летальные исходы).
• Для их профилактики за 1-2 дня до начала ХТ назначают
противогрибковые препараты;
• - с момента начала ХТ начинают неадсорбируемый а/б:
бисептол, офлоксацин, канамицин.
• - поводом для начала комбинированной а/б терапии является
подъем Т тела до 38 градусов, отмеченная 2 или более раз в
сутки, при исключении других причин для ее развития.
53.
• 2 Геморрагический синдром• - выраженная тромбоцитопения – у 1\2 больных
• - летальность от ГС – 5-10% - кровоизлияния в головной мозг,
желудочно-кишечный тракт, легочные кровотечения.
• - Показания для переливания тромбомассы:
• а) при наличии признаков кровотечения на фоне
тромбоцитопении,
• б) при уровне ТР - 30×10.9 + инфекционные осложнения,
• В) при уровне ТР до 10 ×10.9 без признаков инфекции и
повышенной кровоточивости.
54.
• 3 Анемический синдром• - анемия встречается у всех больных ОЛ,
• - основной метод лечения – переливание эритроконцентрата
или отмытых эритроцитов.
• - Показания к переливанию эритромассы:
• - снижение Нв ниже 70 г/л и гематокрита 14-30% ;
• - снижение Нв ниже 45 г/л и Нт менее 14% требует
неотложной гемотрансфузии!
55.
• 4 Энтеропатия• Состояние, характеризующееся поражением слизистой ЖКТ,
возникающее на фоне миелотоксического агранулоцитоза и
проявляющееся болями в животе, вздутием живота, частым жидким
стулом.
• - частота энтеропатии – 10%.
• 5 Синдром лизиса опухоли
• - имеет место при гиперлейкоцитозе более 100тыс и органомегалии.
• -быстрый лизис опухоли выбрасывает в кровь мочевую кислоту и
биогенные амины, блокирующих микроциркуляцию и повреждающих
мембраны клеток, в результате развиваются ОПН, дистресс-синдром,
мозговая кома.
• - больным перед ПХТ проводят гидратацию,
• - назначают аллопуринол, лазикс,
• - с помощью гидроксимочевины снижают лейкоцитоз до 20.000 до
начала ПХТ.
56.
• 6 Нейролейкоз- частота при ОЛЛ – 30-50%; при ОНЛЛ- 5%.
Клинические варианты:
а) менингеальные симптомы – 70%,
б) очаговое поражение головного мозга -5%, в) изолированное
поражение черепно-мозговых нервов – 20%,
• г) полирадикулоневрит – 5%.
• - основной критерий нейролейкоза – повышенный цитоз
спинномозговой жидкости за счет бластоза.
• - развитие нейролейкоза ухудшает прогноз ОЛ.
57.
Современная химиотерапия позволяет получить
полные ремиссии у 50–95% взрослых больных
• В большинстве случаев через 3-4 года
рецидив.
развивается
• 5-летняя
безрецидивная
выживаемость
при
химиотерапии составляет 20–25% для ОМЛ, 30% для
ОЛЛ.
• У больных после лечения острого лейкоза значительно
чаще возникают соматические опухоли.
58.
• Выписка из истории болезни• Николаева Л.Г, 74г,
пенсионерка
Доставлена в НИИ Скорой помощи
июнь 2007г бригадой СП
в связи с учащением приступов стенокардии.
Диагноз: Острый коронарный синдром
59. Жалобы при поступлении:
• - боли в области сердца, сжимающего характера, возникающие приспокойной ходьбе через 100-200м, уменьшающиеся после приема
валидола или нитроглицерина, которые участились до 10 раз в сутки;
- выраженная общая слабость;
- головокружения при попытке выполнить какую- либо нагрузку;
- отсутствие аппетита, снижение настроения, плохой сон.
• Анамнез заболевания: описанные жалобы беспокоят
больную последний месяц, прогрессивно нарастают. К врачам
не обращалась.
• В анамнезе в 2004 году – мастэктомия с последующим курсом
пероральной цитостатической терапии.
60.
• Состояние больной при поступлении средней степенитяжести:
Субфебриллитет;
Астенизирована, несколько заторможена.
Кожа бледная, единичные мелкоточечные геморрагические
высыпания на коже нижних конечностей, синячки разной
степени давности и размеров.
Пастозность голеней.
Периферические лимфоузлы не увеличены.
Пульс 108 в минуту, единичные экстрасистолы
АД 140 и 100 мм рт ст. Тоны сердца приглушены.
Печень + 2,0см; селезенка не увеличена.
Признаки дисциркуляторной энцефалопатии 2 ст.
Грубой очаговой неврологической симптоматики не
выявляется.
Состояние после правосторонней мастэктомии, рубец без
особенностей.
ЭКГ- распространенная субэпикардиальная ишемия в
области левого желудочка.
61.
• Анализ крови клинический:Нв – 65 г/л;
Эритроциты – 2,0 ×1012;
цв.п. – 0,97;
Лейкоциты – 0,45 ×109;
Бласты – 37%;
Промиелоциты – 0%
Миелоциты – 0%
Метамиелоциты – 0%
нейтрофилы палочкоядерные -3 %;
сегментоядерные – 20% = 0,096 ×109;
лимфоциты 30%;
Моноциты – 10%
• тромбоциты - 34 ×109;
• СОЭ - 73 мм/час
• Больная переводится для дальнейшего лечения в гематологическое
отделение Александровской больницы.
62. Клеточный состав костного мозга %
• Клеточный состав костного мозга – 50 В 1 мкл• Мегакариоциты - 0,2-0,6 1/259
• Бласты – 0,1 -1,6%
• Миелобласты –( 0,2-1,7) • Единичные палочки Ауэра
5 7,6 %
Лимфоциты- 4,3%
Промиелоциты – 1,0-4,1
1,2
Моноциты – 0,7-3,1
Миелоциты – 7,0 – 12,22,6 2,6
Метамиелоциты – 8,0- 15,0 2,2
Палочкоядерные – 12,8-23,7 3,4
Сегментоядерные 13,1-24,1 2,8
Всего нейтрофильного ряда- 52,7-68,9 - 12,2%
Эозинофилы – 0,5-5,8
Базофилы – 0-0,5
Эритробласты- 0,2-1,1
Пронормоциты- 0,1-1,2
0,6
Нормоциты базофильные -1,4-4,6 5,4
Нормоциты полихроматофильные 8,9-16,9
Нормоциты оксифильные - 0,8-5,6
Всего клеток эритроидного ряда – 14,5 – 26,5 -- 7,7%
• ЛЭС – 5,1
63.
• 1) Цитохимическое исследование бластныхклеток:
• Реакция на гликоген ++++,
на липиды +
миелопероксидазу 100т
PAS- реакция слабо +
характерно для
клеток миелоидного
ряда
• 2) По данным иммунофенотипирования бластных
клеток выявлена популяция трансформированных клеток с
общим фенотипом CD7, CD13, CD15, CD33, CD34, CD38, HLA-DR,
что более всего соответствует ОМЛ, вариант М1-М2.
• 3) При цитогенетическом исследовании выявлены
множественные хромосомные поломки
64. Диагноз больной Николаевой Л.Г.
• Острый миелобластный лейкоз (вариант М1-М2),вторичный, первый острый период.
• Прогностический индекс – неблагоприятный
• Общесоматический статус менее 50%
• Вторичные острые лейкозы чаще миелобластные.
• Острый миелобластный лейкоз (М1+М2) составляет
50-60% всех случаев ОЛ.
Заболевание чаще встречается у лиц старше 40 лет; характеризуется
быстрым развитием интоксикационного, анемического,
геморрагического синдрома.
Прогноз – неблагоприятный.
65.
• Прогностические факторы при острыхмиелоидных лейкозах:
• 1) Значимые: - возраст более 60 лет – неблагоприятный
прогноз;
- хромосомные нарушения: т(8,12) – высокая частота полных
ремиссий, длительная выживаемость; и другие …
- вторичные лейкозы – прогноз неблагоприятный.
2) Менее значимые: другие FAB – варианты лейкоза (М4-М7)
– неблагоприятный прогноз.;
- общесоматический статус (менее 50% - неблагоприятный
прогноз)
- лейкоциты более 50 в микролитре – благоприятный;
- наличие палочек Ауэра – благоприятный прогноз.
66.
В гематологическом стационаре больной проводится
необходимое обследование для уточнения диагноза и
сопроводительная терапия: переливание тромбо- и
эритромассы, антибактериальная терапия, кардиальная терапия
(нитраты, бетаблокаторы, метаболическая терапия, мочегонные
препараты).
На 5 сутки пребывания в стационаре у больной развивается
тяжелый ангинозный приступ с ЭКГ-картиной острого
распространенного инфаркта миокарда, сложных нарушений
сердечного ритма, от которых больная умирает.
На вскрытии: множественные кровоизлияния во внутренние
органы, в том числе в миокард, перикард с небольшим
количеством крови в перикарде, отек легких.
Посмертное исследование костного мозга подтвердило
диагноз.
67.
Лекциядля 5 курса 4 факультета ФПВ на тему:
« Хронические лейкозы »
Лектор- доцент кафедры ВМТ
Шарова Н.В.
68. Хронические лейкозы – гетерогенная группа опухолевых заболеваний крови, морфологическим субстратом которых является неопластически тра
Хронические лейкозы – гетерогенная группаопухолевых заболеваний крови, морфологическим
субстратом которых является неопластически
трансформированная клетка костного мозга,
сохраняющая определенную способность к
делению, при этом длительно сохраняется
нормальный гемопоэз.
Вторично может происходить метастазирование опухолевых
клеток из костного мозга в периферические органы (лимфоузлы,
печень, селезенка и другие органы)
69. Хронический лимфолейкоз (хронический лимфоцитарный лейкоз - клональное лимфопролиферативное заболевание, первично локализующееся в кост
Хронический лимфолейкоз(хронический лимфоцитарный лейкоз - клональное
лимфопролиферативное заболевание, первично
локализующееся в костном мозге, субстратом которого
являются зрелые лимфоциты.
( Наряду с костным мозгом вторично поражаются лимфоузлы, селезенка,
печень, нервная система и другие органы).
70.
71.
Гемопоэз72. Эпидемиология
• Составляет более 30% всех лейкозов в Северной Америке иЕвропе
Ежегодная заболеваемость в этих регионах составляет 3-4
случая на 100 000 населения, а среди лиц старше 65 лет − 20
на 100 000 населения в год
• Средний возраст к началу заболевания составляет 55 лет,
однако около 10% пациентов моложе 40 лет
• М:Ж = 2:1
В
Европе
и
Северной
Америке
В-клеточный иммунофенотип встречается у 95-98%
больных,
у
2-5%−
Т-клеточный
·
В странах Азии преобладает
Т-клеточный
хронический
лимфолейкоз
73. Этиология ХЛЛ
Для ХЛЛ не доказана этиологическая роль
ионизирующего облучения.
• Для ХЛЛ животных доказана этиологическая роль вирусов.
• Болезнь нередко носит наследственный характер.
• Отмечается увеличение частоты ХЛЛ в семьях с
лимфопролиферативными заболеваниями (ХЛЛ- фактор риска у
родственников 1 поколения)
• .
74.
Патогенез лимфомХЛЛ
Патогенез
СД23,24
СД5
CD5
клеки
Костный
мозг
лимфома
мантийной зоны
В-ХЛЛ
ХЛЛ
СД19
плазматическая
клетка
мантийная зона
множественная
миелома
зародышевый
центр
нативная
В-клетка
В-клетка памяти
Клеткапредшественник
В-клеток
предшественница
лимфопоэза
классическая БХ
лимфоидное
преобладание БХ
фолликулярная
лимфома
лимфома Беркитта
моноцитоидная
В-клеточная лимфома
МALT-лимфомы
ХЛЛ
волосатоклеточный
лейкоз
промиелоцитарный
лейкоз
75.
• Патогенетические особенности ХЛЛ:• - пролиферация клона трансформированных «зрелых»
лимфоцитов;
• в иммунофенотипе - CD19, СД23, СД24, СД5+;
• -характеризуется уменьшением числа мембранных Ig,
• - наличием кластера СД5+, который ассоциируется с
аутоиммунными феноменами (гемолитическая анемия,
тромбоцитопения, тиреоидит..);
• - вытеснение нормального кроветворения с развитием анемии и
тромбоцитопении;
• - в финале болезни может развиваться трансформация ХЛЛ в
лимфосаркому (синдром Рихтера);
• - нарушение противоопухолевого иммунитета часто приводит к
развитию второй опухоли.
76. Клиническая картина
• В70% случаев заболевание начинается постепенно
характеризуется
лишь
абсолютным
лимфоцитозом
периферической крови
и
в
Основные клинические синдромы:
1. Синдром лейкемической пролиферации:
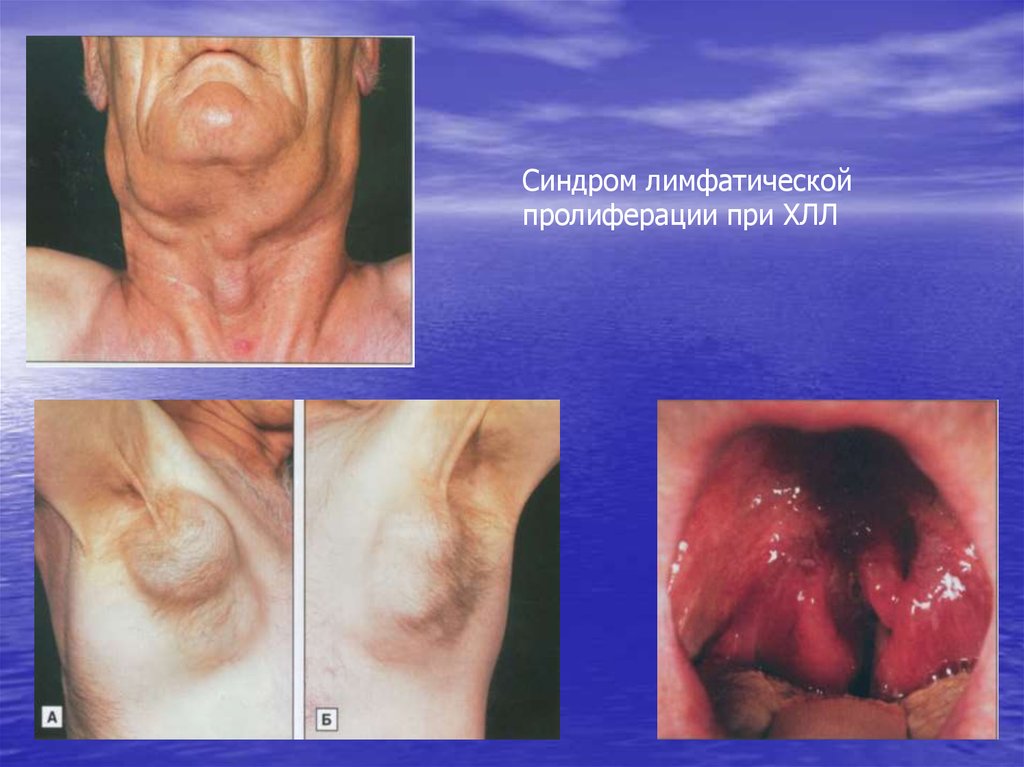
-- Лимфаденопатия:
- периферические лимфатические узлы имеют тестоватую
консистенцию, безболезненные, не спаянные с кожей , клетчаткой
и между собой, не образующие свищей;
• - висцеральные лимфоузлы;
• - позднее появляется увеличение размеров селезенки и печени.
77.
Синдром лимфатическойпролиферации при ХЛЛ
78.
• 2 Симптомы опухолевой интоксикации: истощение, боли вкостях, повышение температуры тела.
• 3 Аутоиммунные осложнения: гемолитическая анемия в
25% и тромбоцитопения в 15%/, возможно сочетание с СКВ.
• 4 Инфекционные осложнения: бронхиты, пневмонии,
сепсис, опоясывающий лишай, выраженная инфильтрация при
укусе насекомых – 70%.
• 5 Возможна трансформация в более агрессивную лимфому
(синдром Рихтера), продолжительность жизни < 6 месяцев
• 30% ХЛЛ имеет индолентное течение.
79. Лабораторные данные
• Лейкоцитоз с абсолютным (более 5 × 109/л)• и относительным лимфоцитозом;
• Морфологически лимфоциты не отличаются от нормальных;
• В ряде случаев встречаются пролимфоциты (обычно менее
10%);
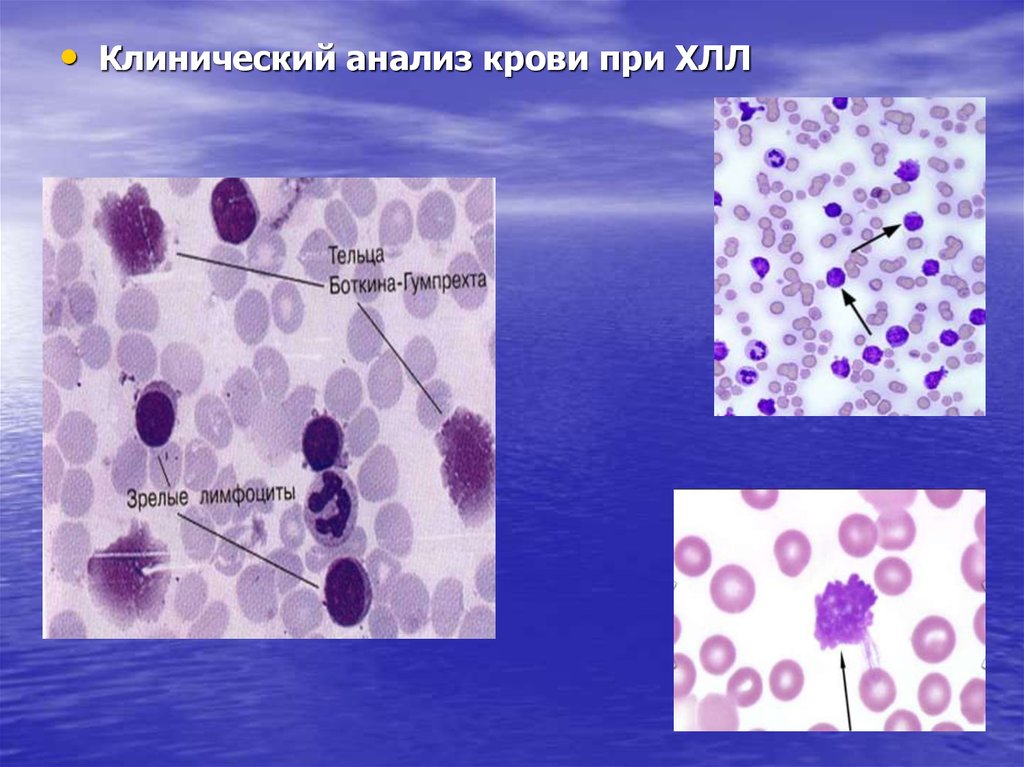
• Характерно наличие в мазке крови клеток Боткина-Гумпрехта
(полуразрушенные ядра лимфоцитов – артефакт, образующийся
при приготовлении мазка);
• Снижение уровня иммуноглобулинов G,A,M.
• По
мере
прогрессирования
тромбоцитопения
развиваются
анемия
и
80.
Нормальные показатели клинического анализа кровиПоказатель
Мужчины
Женщины
Лимфоциты, %
19 37
19 37
Лимфоциты, х 109 /л
1,5 3,5
1,5 3,5
2 10
2 10
0,02 0,8
0,02 0,8
СОЭ, мм/час
1 10
2 15
Ретикулоциты, %о
2 15
2 15
170 350
170 350
Моноциты, %
Моноциты, х 109 /л
Тромбоциты, х 109 /л
81.
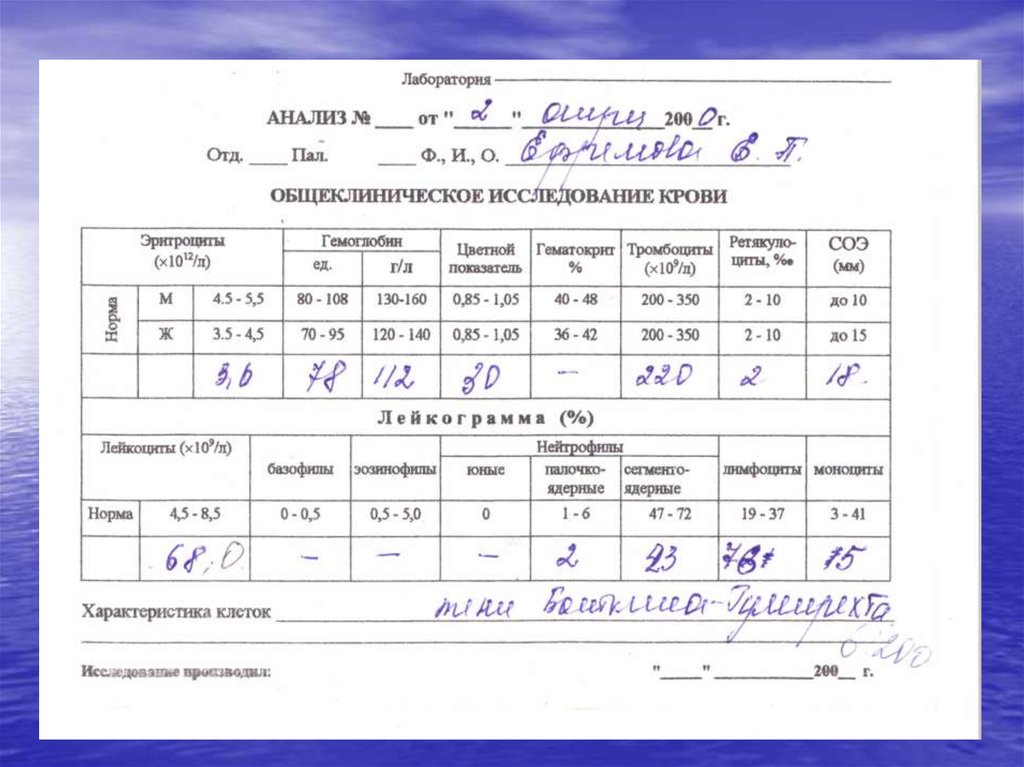
• Клинический анализ крови при ХЛЛ82.
Миелограмма1) При ХЛЛ;
2) нормальная
83.
84. Критерии диагноза
• Абсолютный лимфоцитоз более 5 (10) 109/л• Нормальная
микроскопии
морфология
• Более
30% лимфоцитов
(миелограмме)
лимфоцитов
в
аспирате
при
световой
костного
мозга
• Лимфоидная метаплазия костного мозга в трепанобиоптате.
• Характерный иммунофенотип
(CD19+, CD20+, CD24+, CD5+, CD 23+, слабая экспрессия SIg,
CD79b)
85. Классификация Rai
Классификация RaiСтадия
Характеристика
Медиана
выживаемости
O
Только лимфоцитоз более 15х109/л в
крови, более 40% в костном мозге
Как в популяции15 лет
I
Лимфоцитоз + увеличение
лимфатических узлов
9 лет
II
Лимфоцитоз + спленомегалия и/или
гепатомегалия независимо от
увеличение лимфатических узлов
6 лет
III
Лимфоцитоз + уровень гемоглобина
ниже 110 г/л независимо от увеличения
лимфатических узлов и органов
1, 5 года
IV
Лимфоцитоз + количество тромбоцитов
ниже 100х10 9/л независимо от
увеличения лимфатических узлов и
органов
< 1,5 года
86.
87. Критерии для начала лечения
• (Раноначатое
лечение
не
продолжительность жизни больных !!!)
увеличивает
• Симптомы интоксикации (потливость, снижение веса)
• Анемия или тромбоцитопения в связи с инфильтрацией костного мозга
• Аутоиммунная анемия или тромбоцитопения
• Симптомы компрессии в связи с массивной лимфаденопатией или
спленомегалией
• Удвоение абсолютного лимфоцитоза менее, чем за 12 месяцев
• Лимфоцитоз периферии более 150х109/л .
• Повышенная частота инфекций
• Продвинутая стадия заболевания (C по классификации Binet, III-IV по
классификации Rai)
88. Принципы лечения
• Хлорбутин или циклофосфан• При неэффективности − полихимиотерапия (программы
COP, CHOP и др.)
• Преднизолон при аутоиммунной гемолитической анемии или
тромбоцитопении
• Пуриновые аналоги (флюдарабин по 25 мг/м2) 5 дней
подряд каждые 28 дней
• Анти CD 20 моноклональные антитела (ритуксимаб,
мабтера )
• α-интерферон
• Трансплантация аллогенных стволовых кроветворных
клеток (у молодых пациентов)
89.
• В ряде случаев у больных ХЛЛ используются паллиативныеметоды: лучевая терапия, спленэктомия,
лейкацитоферез.
• Лучевая терапия применяется при наличии выраженной
спленомегалии или конгломерата лимфатических узлов с
признаками компрессии окружающих органов.
• Особое значение придается профилактике и лечению
инфекционных осложнений.
• Больные ХЛЛ должны избегать укусов пчел и комаров.
90.
• Темпы прогрессирования хронического лимфолейкоза широковарьируют: продолжительность жизни колеблется от 2-3 до 30
лет и более
• Средняя продолжительность жизни составляет 5-7 лет
91.
«В течение последних двух десятилетий ХЛЛ из
неизлечимого заболевания превратился в
заболевание, которое в большинстве случаев при
своевременном начале можно успешно лечить,
продлевая жизнь и соматическое благополучие
больных, и которое в настоящее время стало
принципиально излечимым».
92.
93.
• Диагноз: хронический лимфолейкоз, 1стадия по Rai, доброкачественная форма.
• Тактика ведения больного выжидательная.
• С 2005 года у больного рецидивируют простудные
заболевания (3-4 раза в год); появились слабость,
потливость. При очередном осмотре больного размеры
периферических лимфоузлов увеличились до 3-5 см в
диаметре, пальпируется увеличенная селезенка ниже края
левой реберной дуги на 3 см (при УЗИ =15,0см).
В анализе крови лейкоцитоз 80,5 х109/л, 88%
лимфоцитов; гемоглобин – 103г/л, проба Кумбса+.
В трепанобиоптате – пролиферация лимфоцитарного
ростка.
94.
• ДИАГНОЗ: хронический лимфолейкоз, 3 стадия поRai, прогрессирующее течение, аутоиммунная
гемолитическая анемия.
• Начата терапия преднизолоном в дозе 40 мг в сутки.
• Лимфоузлы уменьшились в размерах, количество
лейкоцитов в периферической крови снизилось до
48,5 х109/л, 82% лимфоцитов; гемоглобин –
119г/л
Наблюдается гематологом ЦП ВМА.
95. Хронический миелоидный лейкоз – клональное миелопролиферативное заболевание, характеризующееся -- поражением гемопоэза на уровне стволо
Хронический миелоидныйлейкоз
– клональное миелопролиферативное
заболевание, характеризующееся
-- поражением гемопоэза на уровне стволовой
кроветворной клетки,
--увеличением продукции гранулоцитов
в костном мозге и периферической крови
--и наличием филадельфийской (Ph) хромосомы
и/или химерного гена BCR/ABL.
96. Впервые ХМЛ был описан немецким патологом R.Virchov (1849)- «селезеночная лейкемия» - первый описанный гемобластоз человека. Частота ХМЛ: 1–1,5 слу
Впервые ХМЛ был описан немецкимпатологом R.Virchov (1849)- «селезеночная лейкемия»
- первый описанный гемобластоз человека.
Частота ХМЛ: 1–1,5 случая
на 100 000 населения в год,
постоянна (15–20% всех лейкозов).
Чаще всего встречается
у лиц от 30 до 50 лет.
Соотношение М:Ж= 2:1.
97.
98. Клеточный состав костного мозга %
• Клеточный состав костного мозга – 50 – 250 кл в 1 мкл• Мегакариоциты - 0,2-0,6
• Бласты – 0,1 -1,6%
• Миелобласты – 0,2-1,7
Лимфоциты- 4,3-13,7
Промиелоциты – 1,0-4,1
Миелоциты – 7,0 – 12,2
Метамиелоциты – 8,0- 15,0
Палочкоядерные – 12,8-23,7
Сегментоядерные 13,1-24,1
Всего нейтрофильного ряда- 52,7-68,9
Эозинофилы – 0,5-5,8
Базофилы – 0-0,5
Эритробласты- 0,2-1,1
Пронормоциты- 0,1-1,2
Нормоциты базофильные -1,4-4,6
Нормоциты полихроматофильные 8,9-16,9
Нормоциты оксифильные - 0,8-5,6
Всего клеток эритроидного ряда – 14,5 – 26,5
• ЛЭС – 2,1 – 4,5
Моноциты – 0,7-3,1
99. Этиология
- ионизирующая радиация ( в том числе индуцированнаярентгенотерапией),
- воздействие бензола.
Патогенез
У 95–97% больных ХМЛ во всех опухолевых клетках
выявляется Ph-хромосома (22 аутосома с укороченным
длинным плечом). При этом происходит реципрокная
транслокация 9 и 22 хромосом: t(9;22) (q34; q11), в которой
участвуют 2 клеточных протоонкогена – ABL (Abelson leukemia)
и BCR (breakpoint claster region), расположенных соответственно
на 9 и 22 хромосомах.
На 22 хромосоме образуется химерный ген BCR/ABL,
кодирующий белок Р210, обладающий тирозинкиназной
активностью, что приводит к :
1)
увеличению
пролиферации
миелоидных
клетокпредшественниц с нарушением их дифференцировки;
2) блокаде апоптоза.
3) подавляется нормальное кроветворение.
100. Клинические стадии ХМЛ – этапы лейкозного процесса
• 1 - Хроническая – развернутая (ранняя и поздняя)• 2 - Прогрессирующую (акселерации)
• 3 - Терминальная - бластный криз
101. Хроническая = развернутая фаза ХМЛ
Начальная стадияУ большинства пациентов ХМЛ диагностируется в
хронической стадии, в 40-50% случаев – при отсутствии
каких-либо жалоб - по анализу крови,
- поводом к исследованию анализа крови могла
быть спленомегалия.
102.
Основные симптомы:Признаки
гиперметаболизма
потливость, анорексия);
Спленомегалия
подреберье;
и
ощущения
(снижение
массы
дискомфорта
в
тела,
левом
Нарастание частоты инфекционных заболеваний;
Анемический
синдром (общая слабость, повышенная
утомляемость, одышка при привычной физической нагрузке,
тахикардия, учащение стенокардии);
вторичная подагра;
Геморрагический синдром;
симптомы, обусловленные нарушением микроциркуляции на
фоне гиперлейкоцитоза
103.
У больного с ХМЛувеличение печени,
селезенки, геморрагический
синдром
104.
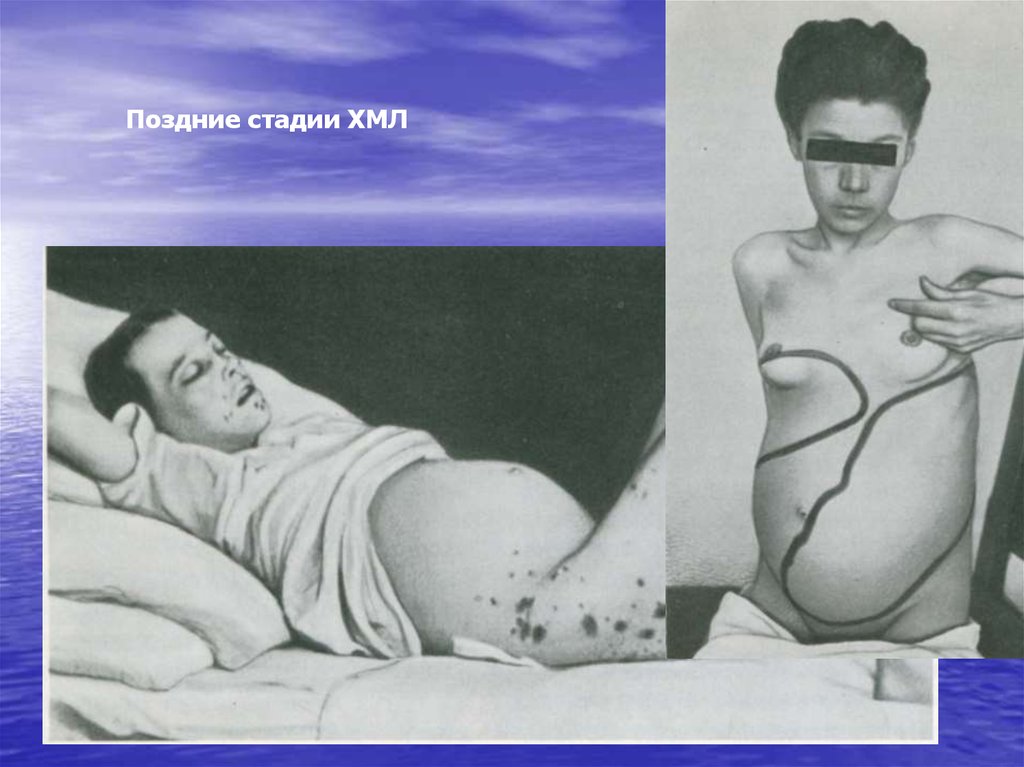
Поздние стадии ХМЛ105. Клинический анализ крови
Лейкоцитоз - в среднем более 50х109/л (возможен и низкийуровень лейкоцитов – 15–20х109/л);
Сдвиг
“влево”
за
счет
палочкоядерных
нейтрофилов,
метамиелоцитов, миелоцитов, редко – промиелоцитов;
Могут выявляться единичные бластные клетки (прогностически
неблагоприятный признак) ;
Характерна эозинофильно-базофильная ассоциация;
В 30% случаев – нормохромная, нормоцитарная анемия легкой
степени;
У 30% больных – тромбоцитоз; реже тромбоцитопения
(неблагоприятный признак)
106. Миелограмма
мозгГиперклеточный костный
Гиперплазия
нейтрофильного
ростка
(лейкоэритробластическое
соотношение достигает
10-20:1)
Количество
клеток
базофильного
и
эозинофильного
рядов
увеличено,
нередко
встречаются
аномальные
формы;
может
быть
повышенным
и
число
мегакариоцитов
76. Костный мозг при хроническом миелолейкозе.
а — в развернутой стадии;
107.
Цитохимическимпризнаком
ХМЛ
являются
снижение
активности щелочной
фосфатазы
нейтрофилов
При
цитогенетическом
исследовании у 95–
97%
больных
выявляется
Ph-хромосома.
При
отсутствии
Ph-хромосомы
молекулярногенетическими
методами
обнаруживается
транслокация
BCR/ABL.
108.
На фоне адекватной химиотерапии практически у всех
пациентов симптомы исчезают:
• - значительно уменьшается в размерах или перестает
определяться селезенка,
• - количество лейкоцитов и лейкоцитарная формула
приближаются к норме (при лечении миелосаном) или
нормализуются
(при
лечении
препаратами
гидроксимочевины).
109. фаза акселерации ХМЛ
В среднем через 3 года, несмотря на продолжающееся
лечение
цитостатиками,
происходит
трансформация
заболевания – опухолевая прогрессия).
У 2/3 пациентов развивается фаза акселерации,
которая характеризуется
прогрессирующей
спленомегалией
с
возможными
инфарктами селезенки,
гепатомегалией;
геморрагическим синдромом,
нарастанием количества гранулоцитов,
нарастанием анемии, тромбоцитоза или тромбоцитопении,
резистентностью к ранее эффективной терапии.
• Продолжительность фазы акселерации составляет 0,5–1
год.
110. Основной лабораторный признак фазы акселерации и бластного криза – прогрессирующее увеличение количества промиелоцитов и бластов в пери
Основной лабораторный признак фазыакселерации и бластного криза – прогрессирующее
увеличение количества промиелоцитов и бластов в
периферической крови и костном мозге.
При цитохимическом исследовании бластов у
70% пациентов определяется миелоидный тип
бластного криза, у 30% – лимфоидный.
111. Терминальная фаза ХМЛ
Обычно терминальная фаза протекает в форме бластногокриза
В редких случаях ХМЛ впервые выявляется в период бластного
криза
Клиника
бластного криза сходна с острым лейкозом. C
наличием большого количества бластов в костном мозге и
определенной клинической картиной.
У 10% пациентов в терминальной стадии развиваются
экстрамедуллярные очаги, состоящие из бластных клеток.
Лабораторные признаки бластного криза при этом могут
отсутствовать (так называемая гранулоцитарная саркома).
Медиана выживаемости больных в этой фазе ХМЛ – 3 месяца
112. Критерии диагноза ХМЛ:
• Лейкоцитоз со сдвигом лейкоцитарной формулы влево• Наличие промежуточных форм нейтрофилов
• Наличие спленомегалии
• Эозинофильно-базофильная ассоциация
• Усиленный миелопоэз в костном мозге
• Низкий уровень щелочной фосфатазы в нейтрофилах
• Обнаружение Ph-хромосомы ( гена BCR|ABL)
113. Дифференциальный диагноз ХМЛ проводится с
-
лейкемоидными реакциями,
острыми нелимфобластными лейкозами,
септическим процессом,
инфекционным (постинфекционным) лейкоцитозом,
агранулоцитозами,
нейтропениями,
инфекционным мононуклеозом,
ВИЧ-инфекцией.
114. Принципы лечения ХМЛ
1.трансплантация аллогенных стволовых
кроветворных клеток
2. Химиотерапия: миелосан (бусульфан),
препаратами гидроксимочевины (литалир, гидреа),
интерферон-α
3. Гливек (иматиниб)
На стадии бластного криза лечение проводится по программе
лечения острых лейкозов.
115. Трансплантация аллогенных стволовых кроветворных клеток при ХМЛ
Аллогеннаямиелотрансплантация в настоящее время
является единственной реальной возможностью излечить
больного ХМЛ.
После
проведения
трансплантации
HLA-совместимого
костного мозга в хронической фазе заболевания
пятилетняя безрецидивная выживаемость отмечается у 50–
70% больных.
При выполнении миелотрансплантации в более далеко
зашедших стадиях ХМЛ результаты заметно хуже (в фазе
акселерации пятилетняя безрецидивная выживаемость <
20%, при бластном кризе – < 10%).
116.
АллопрансплантацияКостного мозга
117. Принципы лечения препаратами гидроксимочевины
Задача врача – снизить и поддерживатьуровень лейкоцитов в пределах 5–15х109/л
118. · Гливек (иматиниб) специфически ингибирует протеинкиназу, продуцируемую химерным геном BCR-ABL, что приводит к блокаде пролиферации и
Гливек (иматиниб)специфически ингибирует протеинкиназу,
продуцируемую химерным геном BCR-ABL, что
приводит к блокаде пролиферации и индукции апоптоза
в Ph-позитивных клетках
·
Гливек показан для лечения больных ХМЛ во
всех фазах болезни.
·
Полная или частичная цитогенетическая ремиссия
достигается у 50% больных в хронической фазе (при
резистентности к интерферону- ), 25% пациентов в
фазе акселерации и 15% – при бластном кризе
·
119.
120. Прогноз при хроническом миелолейкозе
Медианавыживаемости
больных
ХМЛ
стандартной химиотерапии составляет 3,5–4 года.
при
Основные неблагоприятные
прогностические факторы в хронической фазе:
возраст в момент постановки диагноза > 60 лет
наличие бластных клеток в крови (> 3 %) и/или костном мозге (>
5%)
базофилия в крови (> 7 %) и/или костном мозге (> 3%)
тромбоцитоз > 700х109/л.
Развитие фазы акселерации, а тем более бластного криза резко
ухудшает прогноз
121. Истинная полицитемия (эритремия) (болезнь Вакеза, polycythemia vera)
• - доброкачественная опухоль изклеткипредшественницы миелопоэза, сохранившей
способность дифференцироваться по 3
росткам кроветворения, характеризующаяся
повышенной продукцией (неограниченной
пролиферацией) клеток красной крови, а также
лейкоцитов и тромбоцитов.
122.
Частота заболевания – 0,6 на 100 тыс населения.
Преимущественно болезнь людей старше 50 лет.
С одинаковой частотой болеют мужчины и женщины.
У лиц еврейской национальности заболевание встречается в 3 раза
чаще.
• Этиология болезни неясна, среди причин выделяют генетические
факторы.
• Основа патогенеза – хромосомные поломки (в 9
хромосоме), сходные во всей группе хронических
миелопролиферативных заболеваний (JAK2-мутации),
проявляющиеся мутацией гена , приводящая к
клональной пролиферации трансформированной
стволовой клетки с повышенной чувствительностью к
эритропоэтину и неконтролируемому их росту ,
вызывающие:
панмиелоз,
нарушение микроциркуляции,
тромбозы,
геморрагические осложнения.
• .
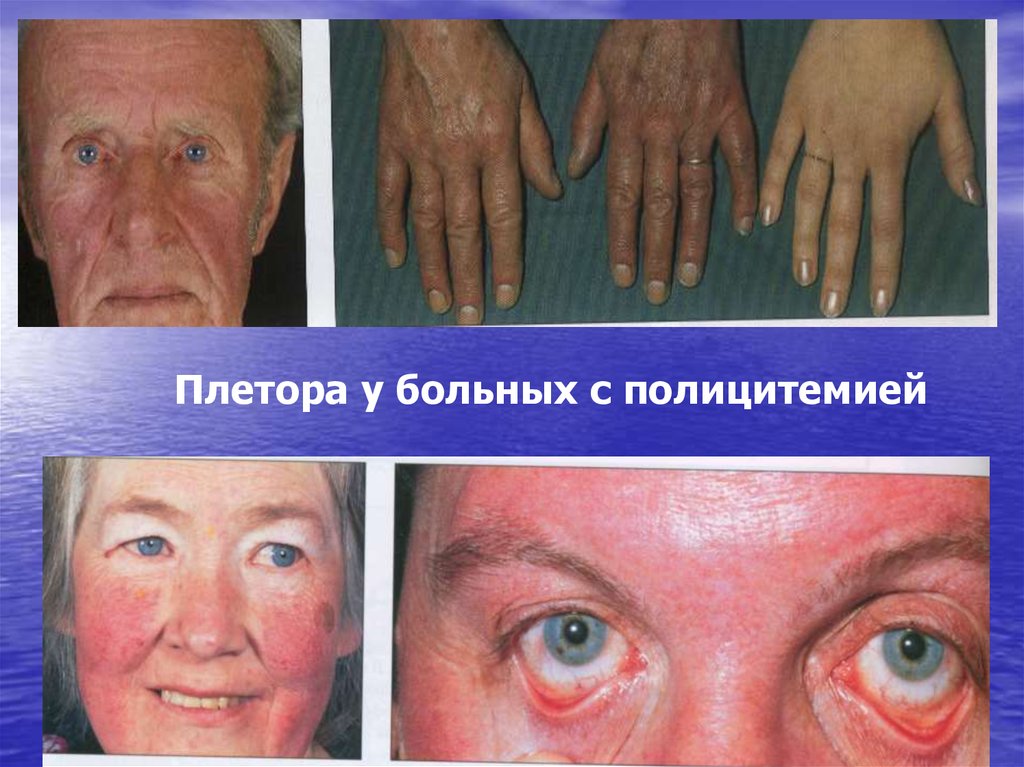
123. Основные клинические синдромы истинной полицитемии
• 1) плеторический синдром;• 2) генерализованный кожный зуд у 50% больных;
эритроцианоз кожи;
3) спленомегалия, особенно выраженная при
вторичном миелофиброзе;
4) тромбозы и тромбэмболии – причина летальных
исходов у 30-40% больных; раннее прогрессирование
ИБС;
5) геморрагический синдром у 30-40% больных
(наклонность к носовым кровотечениям;
кровоточивость десен на фоне лечения зубов);
6) неврологические нарушения в 50-60%
7)поражение периферических сосудов
(эритромелалгия).
124.
Плетора у больных с полицитемией125. Окраска кожи ладоней при эритремии
126.
127.
ЭритремияКожные расчесы
Вторичная
подагра
128.
• СТАДИИ ИСТИННОЙ ПОЛИЦИТЕМИИ:• 1 – начальная: умеренный эритроцитоз, панмиелоз
в костном мозге, небольшое увеличение селезенки.
Длительность – 5лет
2 – пролиферативная: выраженная плетора,
сплено- и гепатомегалия, рецидивирующие
тромбозы, тотальная гиперплазия костного мозга.
3 – анемическая: истощение больных,
нарастающая спленомегалия, нарастает
панцитопения, в костном мозге прогрессирует
постэритремический фиброз.
У 20-30% больных развивается вторичный острый
лейкоз.
Длительность жизни - 10-15 лет.
129. Лабораторные и инструментальные методы
• Анализ крови: повышение гематокрита >55% у М, > 47% у Ж; Нв> 175г/л у М, > 155 г/л у Ж, эритроцитов > 6,0 у М, >5,5
×10.12; лейкоцитоз, тромбоцитоз, снижение СОЭ
до 0 мм/час, дакриоцитоз (каплевидные эритроциты).
Повышение активности щелочной фосфатазы Н.
Увеличение концентрации витамина В12 сыворотки крови.
Миелограмма: 1) в пролиферативной стадии – гиперплазия 3
ростков кроветворения, отсутствие отложения железа, признаки
начинающегося фиброза; 2) в стадии истощения – неэффективный
гемопоэз, фиброз, скопление мегакариоцитов.
130. Лабораторные данные (продолжение)
• Трепанобиопсия (гиперклеточный костный мозг– 60-100%, при сочетании эритроидной
гиперплазии с увеличением мегакариоцитов)
Концентрация эритропоэтина в норме.
рО2 не изменено.
Цитогенетическое исследование – наличие
«филадельфийской» хромосомы или других
хромосомных поломок (9 хромосома)
УЗИ для исключения эритропоэтинсекретирующих
опухолей.
Радиологическое исследование циркулирующей
массы эритроцитов (увеличена).
131. Современные критерии диагностики истинной полицитемии
• А1.Увеличение массы циркулирующих эритроцитов более чем на25% или повышение гемоглобина более 185 г/л для мужчин и 165
г/л для женщин
А2. Отсутствие причин для вторичного эритроцитоза:
- отсутствие семейного эритроцитоза
- отсутствие повышение уровня эритропоэтина вследствие:
а) гипоксии (артериальное рО2 менее 92%);
б) неадекватной продукции опухолью
А3.Спленомегалия.
А4. Клональные цитогенетические аномалии (кроме Ph-хромосомы
или химерного гена BCR/ABL)
А5. Формирование эндогенных эритроидных колоний in vitro
132. Критерии диагностики истинной полицитемии (окончание)
• В1. Тромбоцитоз свыше 400 х 109/л• В2. Количество лейкоцитов свыше 12 х 109/л
• В3. При трепанобиопсии обнаруживается
панмиелоз с
мегакариоцитарной
повышенной
эритроидной
и
пролиферацией
• В4. Низкий уровень эндогенного эритропоэтина.
• В5 Увеличение щелочной фосфатазы нейтрофилов.
Диагноз истинной полицитемии правомочен:
1) при наличии критериев А1+А2 в сочетании с любым
другим критерием категории А;
2) при наличии критериев А1+А2 в сочетании с любыми
двумя критериями категории В.
133.
134.
135. Лечение истинной полицитемии
• Начинают стационарно!!• 1 Уменьшение объема циркулирующей крови с помощью
кровопусканий (250-500мл ч/д) 2-3 раза в неделю и
поддержание Нт на уровне 42-45%, тромбоцитов ниже 400×10.9
2 Профилактика тромбозов – назначение дезаггрегантов
(аспирин, клопидогрель, тромбо-асс, курантил)
3 При гиперурикемии назначение аллопуринола
4 При кожном зуде – антагонисты Н1-Н2рецепторов гистамина,
холестерамин, псорален
5 При необходимости проведения срочных операций –
предварительная флеботомия и эритроцитаферез
136. Лечение истинной полицитемии
• Цитостатики показаны больным пожилого возраста стяжелыми сердечно-сосудистыми осложнениями,
выраженной спленомегалией, лейкоцитозом выше
15000/мкл, тромбоцитозом более 1 000 000/мкл.
Используются гидроксимочевина (гидреа), интерферональфа, анагрелид (преимущественно угнетает
опухолевый рост в мегакариоцитарном ростке), имифос,
миелосан.
• Лечение радиоактивным фосфором.
137.
• Профилактика полицитемии: исключение стрессовыхнагрузок, антиаггреганты, малые дозы цитостатиков.
• Прогноз:
• продолжительность жизни при адекватном лечении
составляет 7-15 лет.
• (при отсутствии лечения – 18 месяцев).
• В 20% случаев – переход в постэритремический
миелофиброз (высвобождение тромбоцитарного
ростового фактора).
• У 20-30% больных ИП в терминальной стадии
развивается острый миелоидный лейкоз
Причины летальных исходов:
- тромбозы и тромбэмболии – 30-40%,
- геморрагические осложнения – 5-10%,
- неврологические расстройства – 60%,
- развитие вторичного острого лейкоза – 20-30%.
138.
Статьярасписания
Наименование болезней,
степень нарушения
функции
Категория годности к военной
службе
болезней
9.
I
графа
II
графа
III
графа
IV
графа
Злокачественны е
новообразования
лимфоид ной, кроветворной
и родственных им тканей
(лейкозы, болезнь
Ход жкина, неход жкинские
лимфомы, злокач ественные
иммунопролиферативные
болезни, множественная
миелома и злокачественные
плазмоклеточные
новообразования
и др.):
а) быстро прогрессирующие,
а также мед ленно
прогрессирующие
со значительными
изменениями
в составе крови и
периодическими
обострениями;
Д
Д
Д
НГ
б) медленно
прогрессирующие
с умеренным нарушением
функции кроветворной
системы и редкими
обострениями;
Д
Д
В
НГ
в) состояние пос ле
завершения луч евой или
цитостатической терапии
Д
Д
Г
НГ
139.
• К пункту «а» относятся злокачественные заболеваниялимфоидной, кроветворной и родственных им тканей, когда
эффект от проводимого лечения отсутствует или носит
временный характер.
• К пункту «б» относятся заболевания с медленно
прогрессирующим течением, длительным положительным
эффектом от лечения, частотой обострений, не превышающих 1
раза в год, с сохранением способности исполнять обязанности
военной службы.
• К пункту «в» относятся состояния после лучевой и
цитостатической терапии по поводу злокачественных
заболеваний. Заключение о необходимости предоставления
отпуска по болезни выносится после первого курса лучевой или
цитостатической терапии.
• Военнослужащим, проходящим военную службу по контракту и
освидетельствующимся в связи с увольнением в запас,
заключение выносится по пункту «а», независимо от
локализации, стадии и распространенности злок процесса и
времени его начала.