


















































































































 Химия
ХимияПохожие презентации:










Физическая химия. Химическая термодинамика. Раздел №1
1. Физическая химия. Раздел №1. Химическая термодинамика
Литература:1.
2.
3.
4.
5.
Я.И.Герасимов. Курс физической химии (т.1)
П. Эткинс. Физическая химия (т.1)
М.Х. Карапетьянц. Химическая термодинамика
Е.Н. Еремин. Основы химической
термодинамики
Solomonov B., Mukhametzyanov T. Encyclopedia of
Physical Organic Chemistry, 2017, v.1, p.315-368.
2. Газовые законы
Идеальный газ (отсутствуют межмолекулярные взаимодействия иразмеры молекул газа намного меньше расстояний между ними)
P× V = n× RT
̶ идеальный газовый закон
P – давление, V – объем, n – число
молей,
R – газовая постоянная, T - температура
Объем газа – объем сосуда, где находится газ
Давление газа
F
внешнее
A
F – сила, A – площадь
P=
внутреннее
(газ)
F
A
При равновесии
Pвнеш = Pвнутр
3. Температура – определяет направление потока энергии
Анизкая t
B
А
B
высокая t
равные t
шкала Цельсия (0°С – tпл лед; 100°С – tкип H2O)
шкала Кельвина (0°С – -273,15°С)
P
Закон Бойля: V ~
T(Кельвин) = t(Цельсий) + 273,15
m
n – число молей
M
t
V
1
P
4.
VA
B
Закон Гей-Люссака: V~T
V = V0 × (1+ α × t)
C
V0 – объем газа при t=0°C
α – коэффициент терм. расширения
-273.15
t°C
Принцип Авогадро: равные объемы газов содержат
одинаковое число молекул (t и P = const)
Смесь газов: A и B газы
nA RT
nBRT
pA =
; pB =
V
V
nA
nB
yA =
; yB =
;
nA + nB
nA + nB
Закон Дальтона:
pA = y A Pобщ;
pA, pB – парциальные давления
yA, yB – мольные доли
pB = yBPобщ
Давление, создаваемое смесью идеальных газов, представляет собой сумму
давлений, создаваемых каждым газом, занимающим тот же объем
в одиночестве
5.
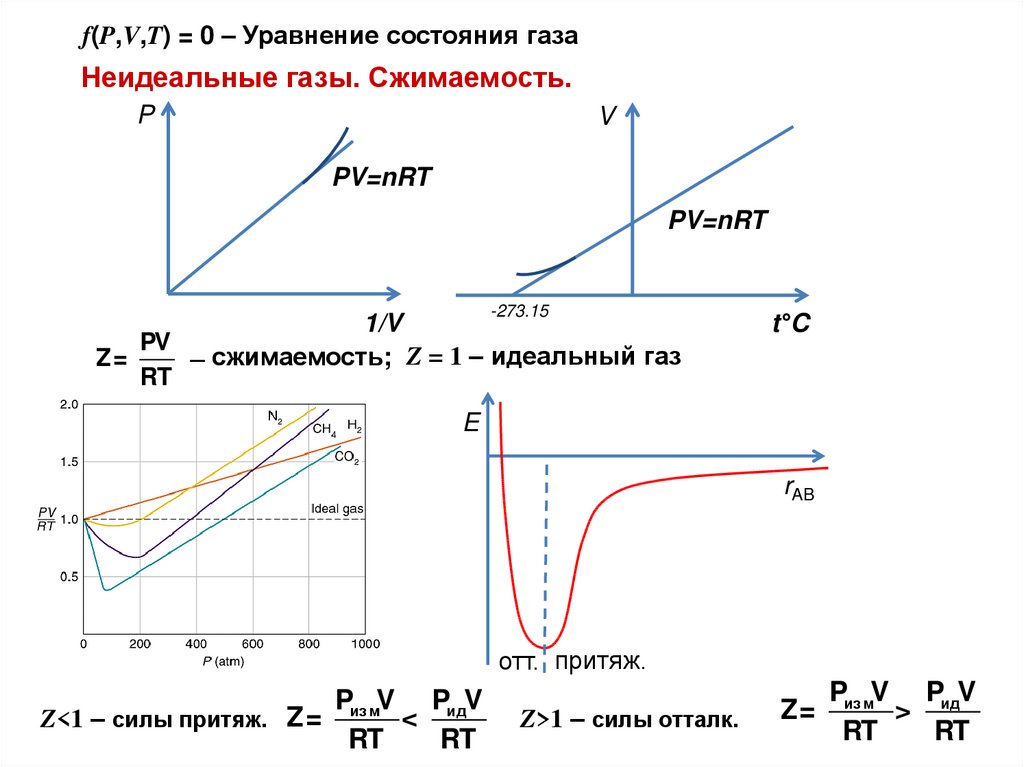
f(P,V,T) = 0 – Уравнение состояния газаНеидеальные газы. Сжимаемость.
P
V
PV=nRT
PV=nRT
-273.15
PV
Z=
RT
1/V
̶ сжимаемость; Z = 1 – идеальный газ
t°C
E
rAB
отт. притяж.
P V P V
Z<1 – силы притяж. Z= из м < ид
RT
RT
Z>1 – силы отталк.
Z=
Pиз мV PидV
>
RT
RT
6.
Вириальное уравнениеB
C
Z=
1+
+
+ ...
2
Z= 1+ B'× P; Z= 1+ B'× P+ C'× P + D'× P ;
V V
2
3
B
C - B2
B' =
; C'=
; B, B' = f(T) ̶ вириальные коэффициенты
2
(RT)
RT
B и B’ = 0
(при T – температуре Бойля все газы ведут себя
подобно идеальному газу)
B>>C>D
B,C и D – зависят от температуры и это недостаток
Уравнение Ван-дер-Ваальса (vW)
a
P+
Vm - b = RT
2
Vm
PV=RT
b ~ собственный объем молекулы; P(идеал) = P(неидеал) + P’
a ~ силы притяжения между молекулами
a и b не зависят от T
7.
Коэффициенты Ван-дер-Ваальса и 2ой вириальный коэффициентRT
a
P=
- 2
Vm - b Vm
PVm
Vm
a
1
a
=
=
RT
Vm - b RT× Vm 1- b RT Vm
Vm
b/Vm <<1 (при средних давлениях)
(vW) Z=
1
b
b2
= 1+
+ 2 + ...
1- b Vm
Vm Vm
a
RT
a
b=
B= 0
RT
b× RT> a (при высоких T)
RT a
P=
- 2
Vm -b Vm
(vW)
B C
+ 2 + ...
V V
a 1
b2
Z= 1+ b+ 2
×
RT Vm Vm
1
a
Z=
1- b Vm RT× Vm
B= b -
Z= 1+
Т-ра Бойля
b× RT< a (при низких T)
R
P
=
;
T V Vm - b
a
P
T×
P=
T
Vm2
V
a и b находят из данных по P и T при V = const
8.
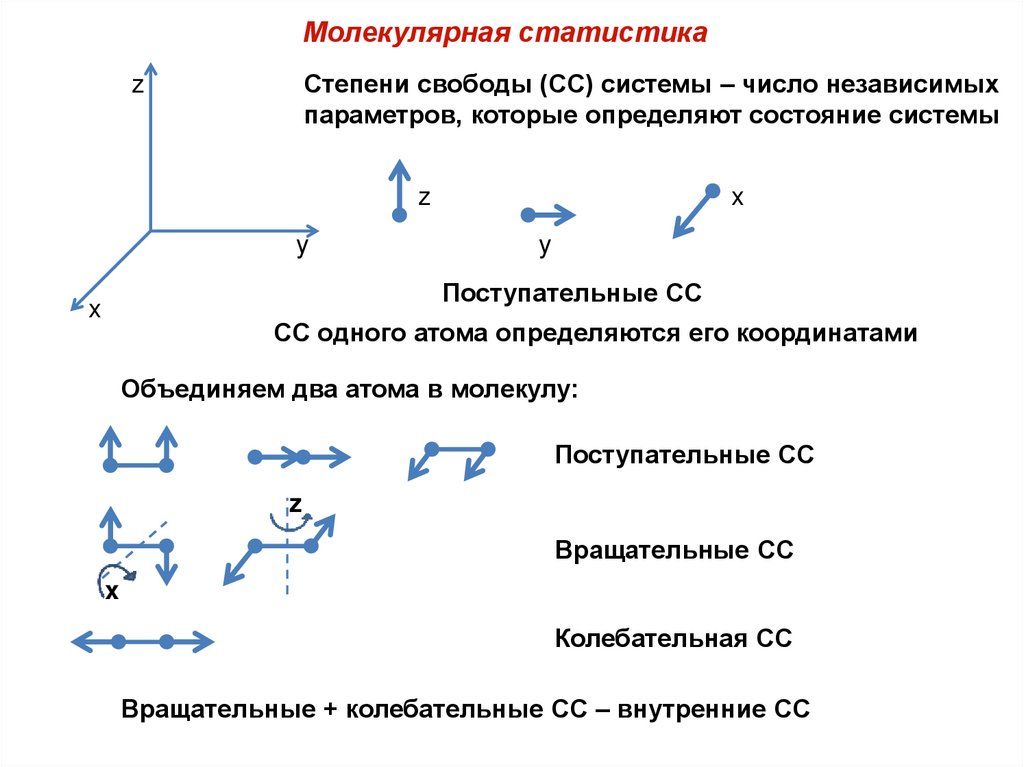
Молекулярная статистикаСтепени свободы (СС) системы – число независимых
параметров, которые определяют состояние системы
z
z
y
x
y
Поступательные СС
x
СС одного атома определяются его координатами
Объединяем два атома в молекулу:
Поступательные СС
z
Вращательные СС
x
Колебательная СС
Вращательные + колебательные СС – внутренние СС
9.
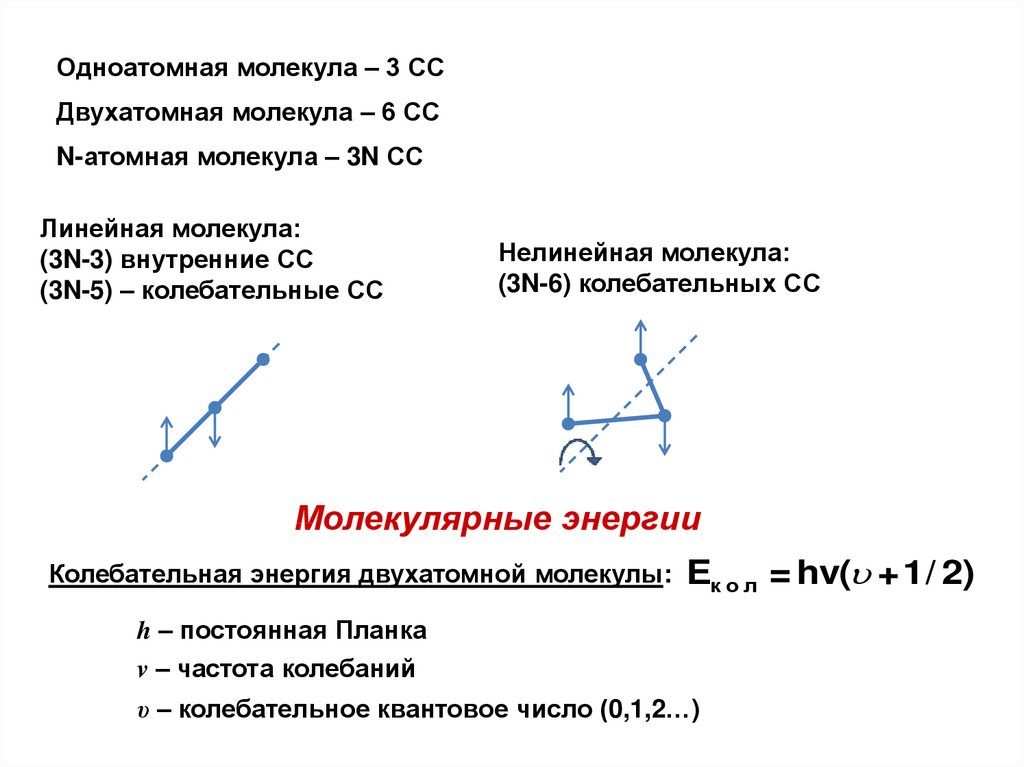
Одноатомная молекула – 3 ССДвухатомная молекула – 6 СС
N-атомная молекула – 3N СС
Линейная молекула:
(3N-3) внутренние СС
(3N-5) – колебательные СС
Нелинейная молекула:
(3N-6) колебательных СС
Молекулярные энергии
Колебательная энергия двухатомной молекулы:
Eк о л = hv( +1/ 2)
h – постоянная Планка
v – частота колебаний
υ – колебательное квантовое число (0,1,2…)
10.
Вращательная энергия двухатомной молекулы А-В:2
Eвр
h2
=
( J +1)J
2
8π I
I – момент инерции I= mr r (r – расстояние А-В)
m1m2
mr =
– приведенная масса
m1 + m2
J – вращательное квантовое число (0,1,2…)
Поступательная энергия:
E пос т
n2h2
=
8m×l2
m – масса молекулы
n –поступательное квантовое число (0,1,2…)
l
Пример: He, l =10 см
Средняя кинетическая энергия E = 2.1·10-21 Дж
n= ( 8m× E)
ΔE → 0
1/ 2
l
× = 1.6× 10 9
h
11.
9ε3
9
ε'6
6.25
ε2
4
4
ε1
1
1
l
2l
2.25
ε'4
ε'3
ε'2
ε'1
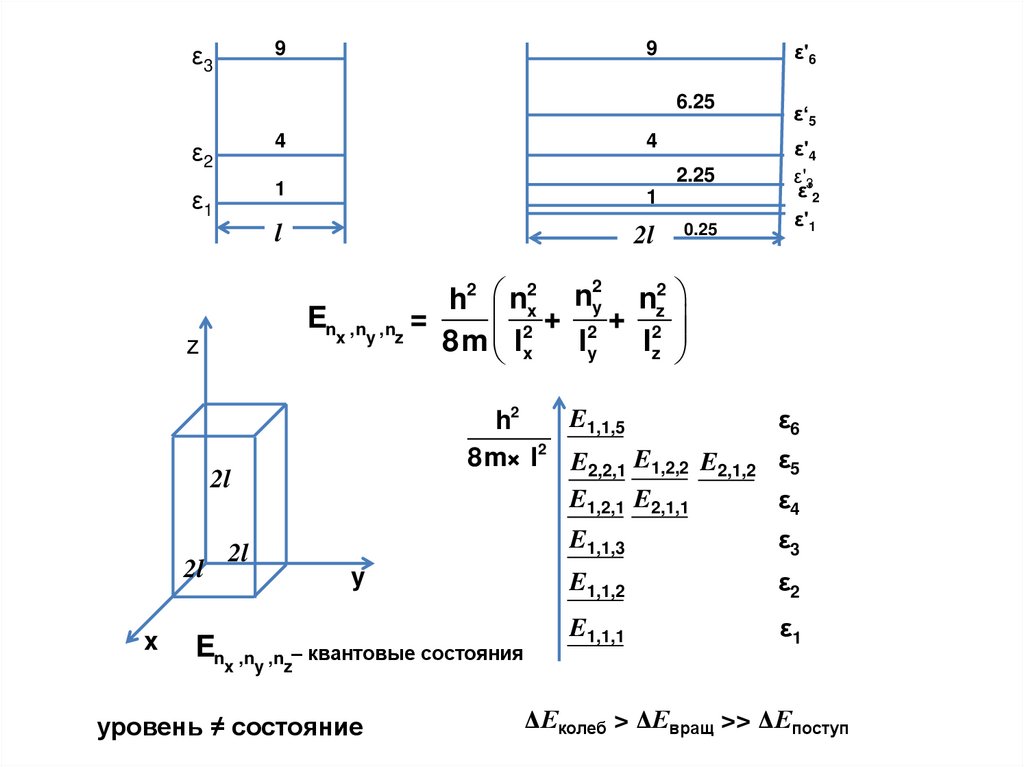
2
h2 n2x ny nz2
En ,n ,n =
2 + 2 + 2
x y z
8m lx
ly
lz
z
E1,1,5
ε6
h2
8m× l2 E2,2,1 E1,2,2 E2,1,2 ε5
E1,2,1 E2,1,1
ε4
E1,1,3
ε3
2l
2l
2l
x
0.25
ε‘5
y
En
– квантовые состояния
E1,1,2
ε2
E1,1,1
ε1
x ,ny ,nz
уровень ≠ состояние
ΔЕколеб > ΔЕвращ >> ΔЕпоступ
12. Проблема
1. Многие эксперименты в физической химиивключают макроскопические системы с
огромным числом частиц.
2. Не все молекулы в данной системе имеют
одинаковую энергию и занимают
различные энергетические уровни.
Вопросы:
1. Как молекулы распределяются по
энергетическим уровням.
2. Какова доля молекул занимающих данный
энергетический уровень.
13.
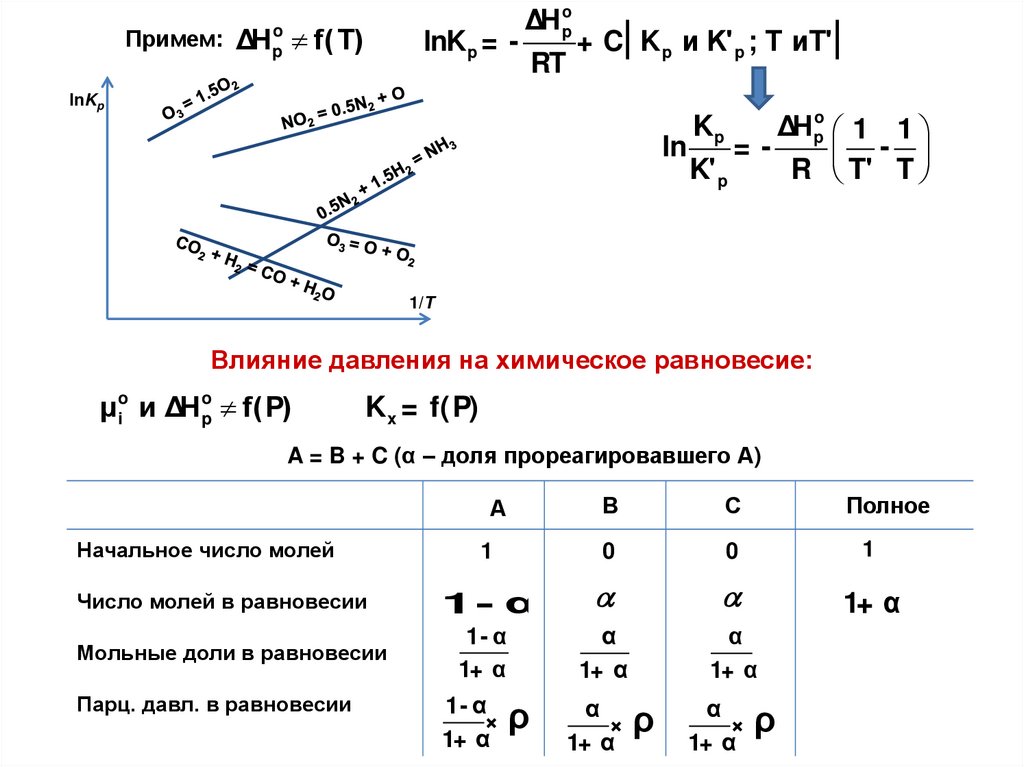
Двухатомная молекулаПримем:
Микросостояния
1
3
5
E
:
hν
,
hν
,
hν
Eк ол= hν( + 1/ 2) к ол
2
E кол
микросостояние 1
микросостояние 2
А
В
Распределения
микросостояние 3
А,В
В
А
1
Распределения
3 hν
2 hν
1 hν
0 hν
ΣEк ол= 2hν
2 молекулы (A и B)
2 hν
1 hν
0 hν
E кол
2
( 3)
E(к1)ол= 0 hν E(2)
к ол= 1hν Eк ол= 2 hν
Система 1:
Система 2:
2
2
ΣEк ол= 3hν
3 молекулы (A, В и С)
1
2
3
А
В
С
ВС
АС
1
АВ
4
5
6
7
8
9
10
А
В
С
В
А
С
С
А
В
А
С
В
В
С
А
С
В
А
А,В,С
2
3
14.
N!N –общее число молекул в системе
W(число макросостояний)=
N 0 !N 1!N 2!...N i! Ni –число молекул на i-уровне
W1 =
3!
= 3
2!× 1!
Система 3:
W2 =
3!
=6
1!× 1!× 1!
1000 молекул
E кол
999!× 1!
(0!=1)
ΣEк ол= 3 hν
1
Распределения
1000!
3!
=1
3!
Число молекул
3hν
2hν
1hν
0hν
W1 =
W3 =
= 1000 W2 =
999
1
1000!
998!× 1 !× 1!
1
1
998
2
5
= 9.9 9× 10 W3 =
3
997
3
1000!
= 1.66167 × 10
8
997!× 3 !
1) W3 – наиболее вероятное распределение (99,9%); ΣW i= 1,67157× 10 8
2) n↑, W→∞
15.
Закон распределения Больцмана (1871)В точке max f(x) – f(x+dx)=df~0 (dx→0)
E
2(Ej - Ei ) = Ek - Ei
Ni~f(Ei)?
Ek, Nk
Ej, Nj
Ei, Ni
1 мол.
2 мол.
WН В – наиболее вероятное распределение
N!
WН В =
...N!N
i
j!Nk!...
N!
W'=
...(N i + 2)!(N j - 3)!(N k + 1)!...
в т. max W
НВ
=W’→dW=0
при
(E = const)
E2, N2
N i!N j!N k!= ( N i + 2) !( N j - 3) !( N k + 1) !
E1, N1
N j!(N j -1)!(N j - 2)!= (N i + 2)!(N i + 1)!(N k + 1)!
N k + 1 N k ; N j -1 N j - 2 N j ; N i + 1 N i + 2 N i ;
N j Ni
=
N k N j
2
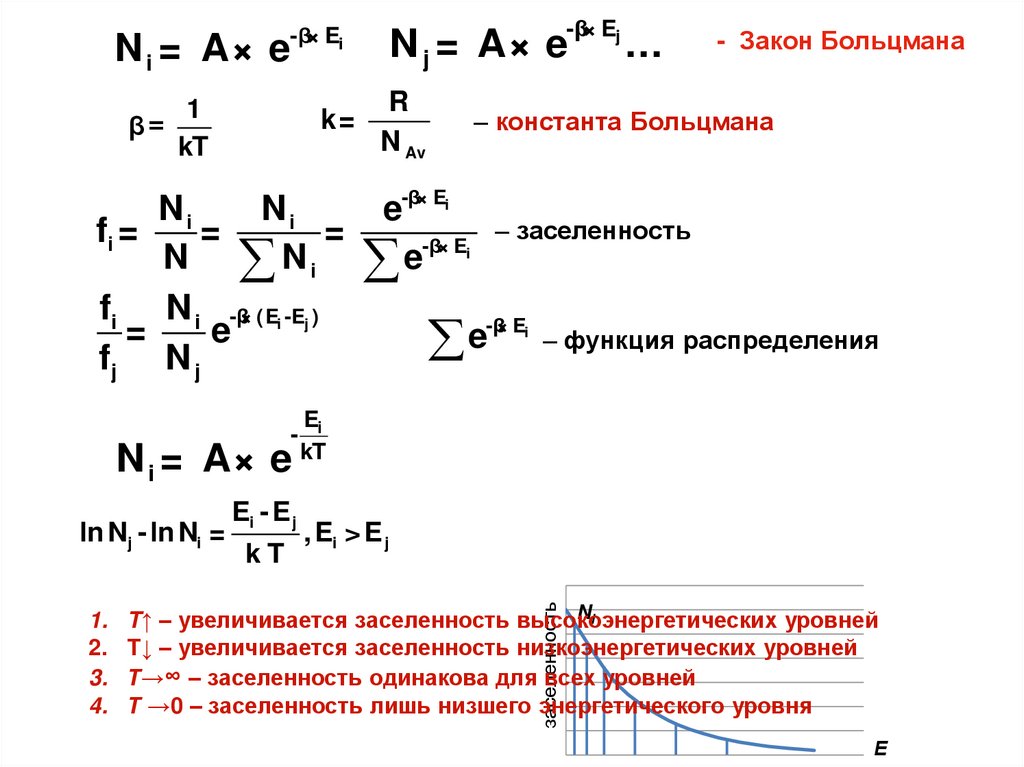
16.
-β× Ej-β× Ei
N j = A× e
N i = A× e
k=
Ni
Ni
fi =
=
=
N Ni
fi N i -β× (Ei -Ej )
=
e
fj N j
-
N i = A× e
ln Nj - ln Ni =
1.
2.
3.
4.
Ei - E j
kT
R
N Av
- Закон Больцмана
– константа Больцмана
e-β× Ei
-β× Ei
e
– заселенность
-β× Ei
e
– функция распределения
Ei
kT
, Ei > E j
заселенность
1
β=
kT
...
Ni
T↑ – увеличивается заселенность высокоэнергетических
уровней
Т↓ – увеличивается заселенность низкоэнергетических уровней
Т→∞ – заселенность одинакова для всех уровней
Т →0 – заселенность лишь низшего энергетического уровня
Е
17.
IClυ
0
1
2
3
4
5
заселенность
100
53,5
28,6
15,6
8,2
4,4
ν=1,15х1013
E0
=
сек-1
h=6,626х10-34
Дж х сек Т – ?
Ni = A e
-β × E
i
0 , E1 = hν = ( 1, 15× 10 13 с-1 ) × ( 6, 626 × 10 -34 Дж× с) = 7, 62 × 10 -21 Дж
E2 = 2E1
[ Eк ол= hν( + 1/ 2)]
E3 = 3E1 , E4 = 4E1 , E5 = 5E1
-(3E1 - E1 )
-2E1 N i – относительная заселенность
N3
= exp
= exp
Nj
N1
kT
kT
-2E1
T=
k=1,381х10-23 Дж·К-1 ln(N3/N1)=-1,25
k ln N 3 N 1
T= 863К
Если система находится в равновесии, любая пара из N дает одну T.
Геометрическая прогрессия это численная последовательность, каждое из чисел равняется предыдущему,
умноженному на определенное постоянное число q для данной прогрессии,
которое называется знаменателем геометрической прогрессии.
Арифметическая прогрессия это численная последовательность в которой все члены получаются из
предыдущего методом добавления к нему 1-го и того же числа d, которое называется разностью арифметической
прогрессии.
18.
Равновесная термодинамика (ТД)система
окружение
I
II
III
I – открытые системы (обмен энергией и веществом)
II – закрытые системы (нет обмена веществом)
III – изолированные системы (нет обмена ни веществом, ни энергией)
n, P, V, T – основные ТД параметры
ТД параметры: 1. экстенсивные – V
2. интенсивные – P, T
Равновесное состояние: ТД параметры (система) = ТД параметры (окружение)
P1, V1, T1
ТД процесс
P2, V2, T2
19.
Термодинамический процесс1.Самопроизвольный.
2.Несамопроизвольный
3.Изохорный (V=const)
4.Изобарный (P=const)
5.Изотермический (T=const)
Нулевой Закон термодинамики
Две системы, находящиеся в термическом равновесии с
третьей системой, состоят в термическом равновесии друг с
другом.
Первый Закон термодинамики
ΔU = q + w
ΔU – изменение энергии, q – теплота, w – работа
Формулировки Первого Закона:
1)Энергия изолированной системы остается постоянной.
2)Вечный двигатель I-го рода невозможен.
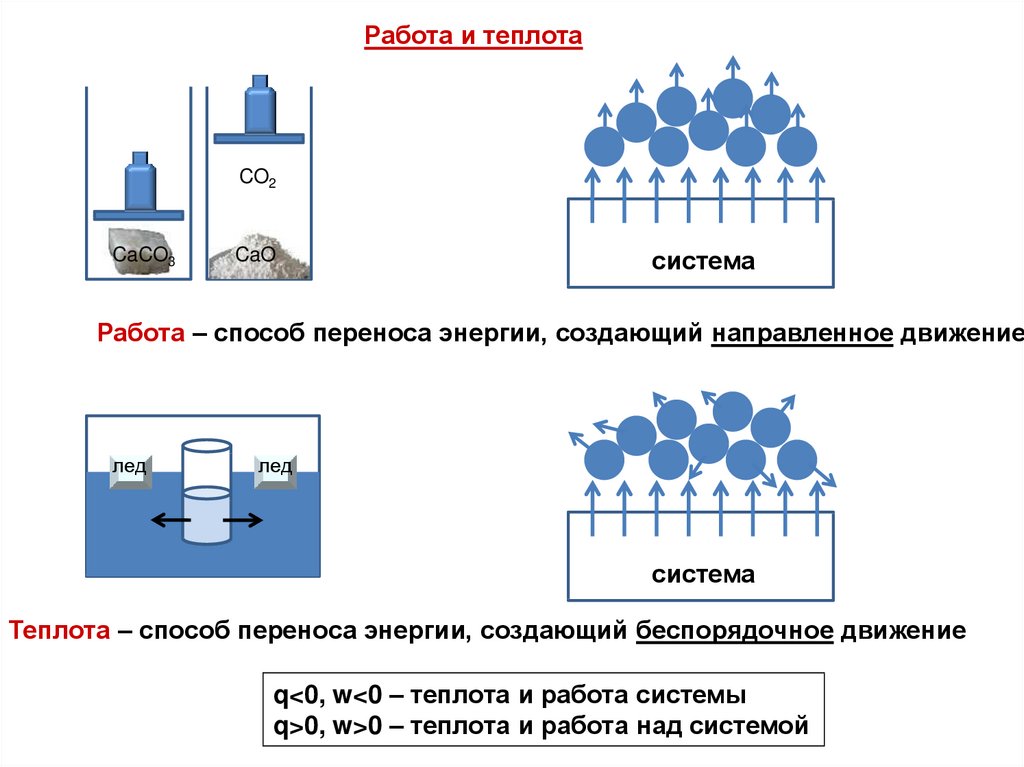
20.
Работа и теплотаCO2
CaCO3
CaO
система
Работа – способ переноса энергии, создающий направленное движение
лед
лед
система
Теплота – способ переноса энергии, создающий беспорядочное движение
q<0, w<0 – теплота и работа системы
q>0, w>0 – теплота и работа над системой
21.
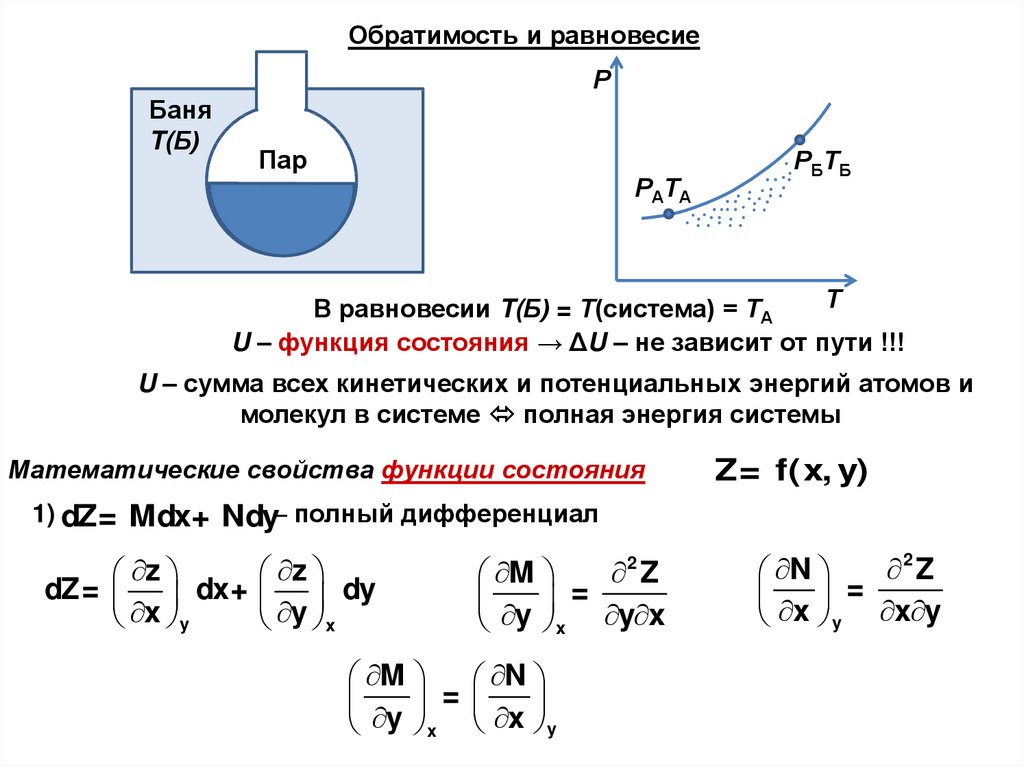
Обратимость и равновесиеР
Баня
T(Б)
Пар
РАТА
РБТБ
Т
В равновесии T(Б) = Т(система) = ТА
U – функция состояния → ΔU – не зависит от пути !!!
U – сумма всех кинетических и потенциальных энергий атомов и
молекул в системе полная энергия системы
Математические свойства функции состояния
Z= f(x, y)
1) dZ= Mdx+ Ndy– полный дифференциал
z
dZ= dx+
x y
z
y dy
x
2 Z
M
y = y x
x
M
N
=
y
x x y
2 Z
N
=
x y x y
22.
22) Z - Z =
2
1
dZ= df( x, y) = f( x
1
3)
2
2
, y 2 ) - f( x1 , y1 )
1
dZ= 0
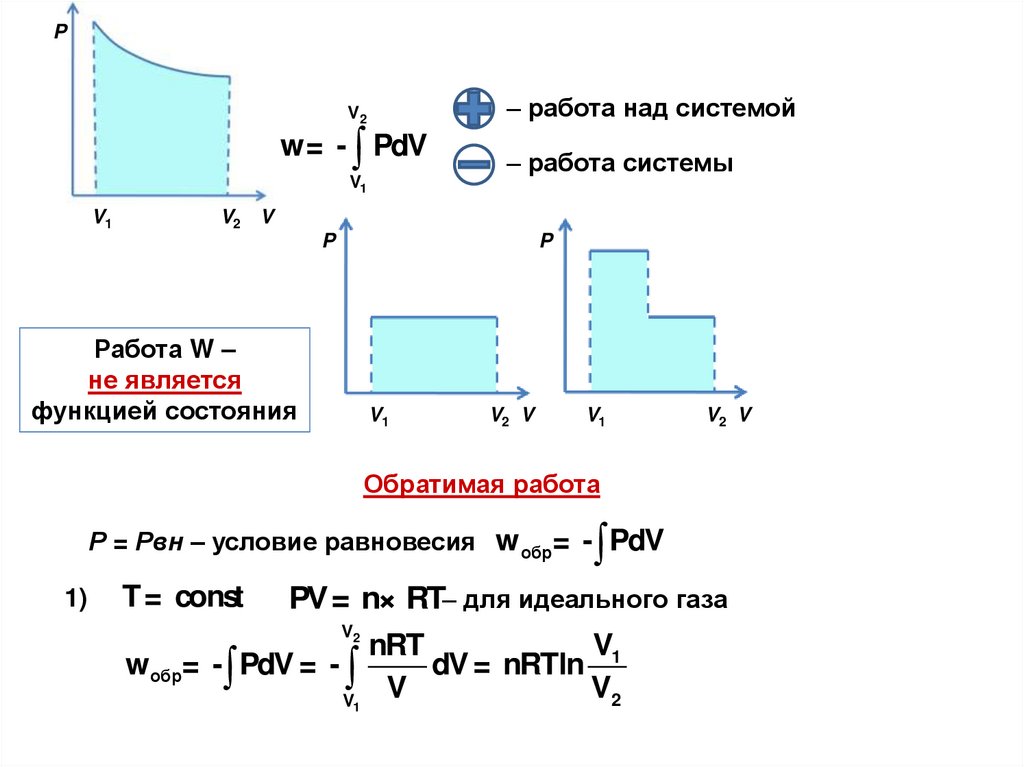
Механическая работа
w= - fdx
f – сила, dx – величина перемещения
f
w= - A dx= - PвнdV
A
w= -Pвн dV = -Pвн× ΔV Pвн f( V)
Pвн
ΔV = Vк он - Vнач
dx
при Pвн = const
A
23.
PV2
w= - PdV
V1
V1
– работа над системой
– работа системы
V2 V
P
P
Работа W –
не является
функцией состояния
V1
V2 V
V1
V2 V
Обратимая работа
Р = Рвн – условие равновесия w обр= - PdV
1)
T= const
PV = n× RT– для идеального газа
V2
V
nRT
dV = nRTln 1
V
V2
V1
w обр= - PdV = -
24.
Для обратимой работы Р = Рвнwобр PdV
1) Процесс расширения:
Vb Va
Pвн P P
w max , при δP→0
2) Процесс сжатия:
Va Vb
wобр w (δP=0)
a b
Pвн P P
w min , при δP→0
w ( P P)dV
w ( P P)dV
wобр w (δP=0)
b a
Пример: испарение жидкости (P=const)
w PdV P(Vпар Vж ) P Vпар ; (Vпар Vж )
Пар: PV=nRT
w n RT
25.
ТеплоемкостьΔ U= q- PвнdV
dV = 0 – процесс изохорный
Δ U= qV
δqV
– истинная теплоемкость
C V = lim
δT 0 δT
ΔqV
– средняя теплоемкость
CV =
ΔT
U
CV =
ΔU= CV dT
T V
Δ T=
1
q
C
V
Размерность: Дж/К, Дж/К·моль (мольная), Дж/К·г (удельная)
CV=f(T);
при низких
CV→0
при T→0
T C V = A× T 3
C V = a+ b× T+ c× T 2 + ...
CV = a'+ b'× T -1 + c'× T -2 + ...
a’, a, b’, b, c’, c, – эмпирические константы
26.
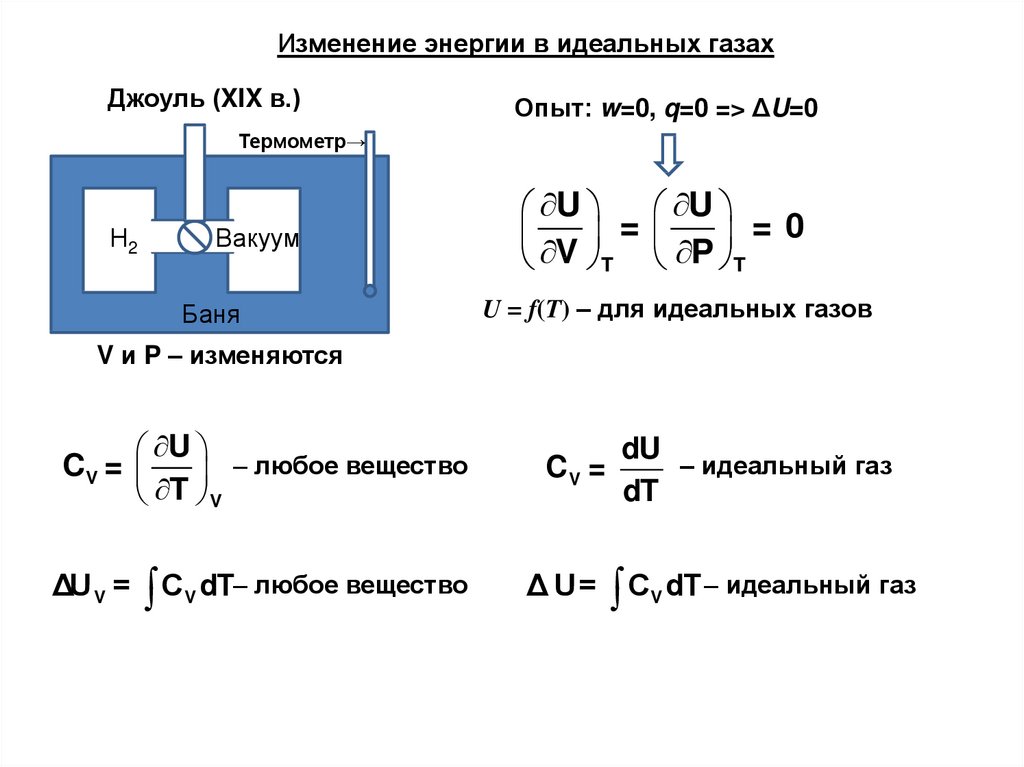
Изменение энергии в идеальных газахДжоуль (XIX в.)
Опыт: w=0, q=0 => ΔU=0
Термометр→
Н2
Вакуум
Баня
U
U
=
= 0
V T P T
U = f(T) – для идеальных газов
V и P – изменяются
U
CV =
– любое вещество
T V
ΔU V =
C
V
dT– любое вещество
CV =
Δ U=
dU
– идеальный газ
dT
C
V
dT – идеальный газ
27.
ЭнтальпияΔ U= q P - Pвн dV
q P= Δ U+ Pвн dV (Pвн= const)
Δ H= Δ U+ Δ PV
H= U+ PV
Δ H= Δ U+ P× Δ V
(P= const)
Δ H= q P - Pвн dV+ P× ΔV
Для обратимых процессов
Pвн= P
Δ H= q p
Pвн× dV = Pвн× ΔV = P× ΔV
H – функция состояния
H
CP =
T
P
Δ H=
C dT
P
28.
Докажем: CP – CV = nR для идеальных газовH= U+ PV
H U PV
=
+
T P T P T P
dU
U U
=
=
= CV
T P T V dT
PV
V
= P×
= nR
T P
T P
PV= nRT
CP - CV = nR
H
= CP
T P
Изменение энтальпии в идеальных газах
H1 = U1 + P1V1
T1 = T2
H 2 = U 2 + P2 V2
P1 V1 = P2 V2
U1 = U 2
(процесс изотермический)
P1 V1 P2 V2
=
T
T
1
2
H1 = H 2
29.
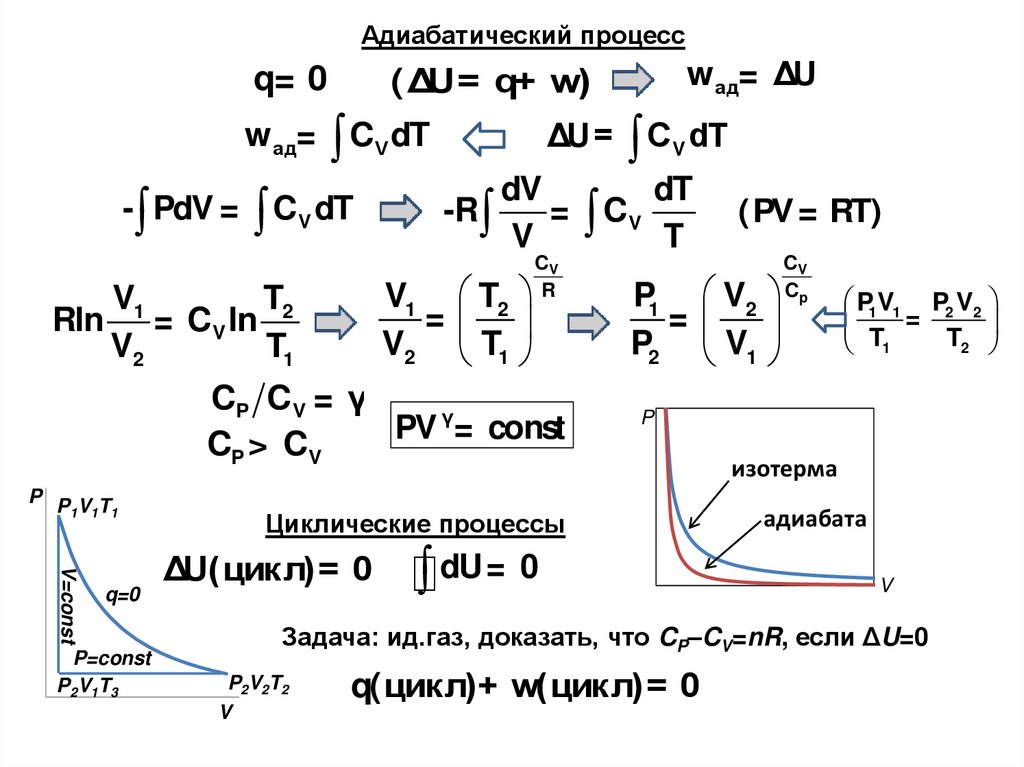
Адиабатический процессw ад=
- PdV =
C
CV dT
V
ΔU=
dT
dV
-R
=
V
V1 T2
=
V2 T1
V1
T2
Rln = CV ln
V2
T1
CV
R
CP CV = γ
PV γ= const
CP > CV
P PVT
1 1 1
V=const
q=0
P=const
P2V1T3
wад= ΔU
(ΔU= q+ w)
q= 0
C
V
dT
dT
CV T
P1 V2
=
P2 V1
CV
Cp
P1 V1 P2 V2
=
T
T
1
2
P
Циклические процессы
ΔU(цик л) = 0
(PV = RT)
dU= 0
изотерма
адиабата
V
Задача: ид.газ, доказать, что CP–CV=nR, если ΔU=0
P2V2T2
V
q(цик л)+ w(цик л) = 0
30.
w= CV (T2 - T1 ) - P2 (V1 - V2 )+ 0 = CV (T2 - T1) - nR(T3 - T1)q= 0+ Cp ( T3 - T2 )+ C V ( T1 - T3 )
w+ q= 0 = Cp ( T3 - T2 )+ C V ( T2 - T3 ) - nR( T3 - T2 )
Cp - C V = nR
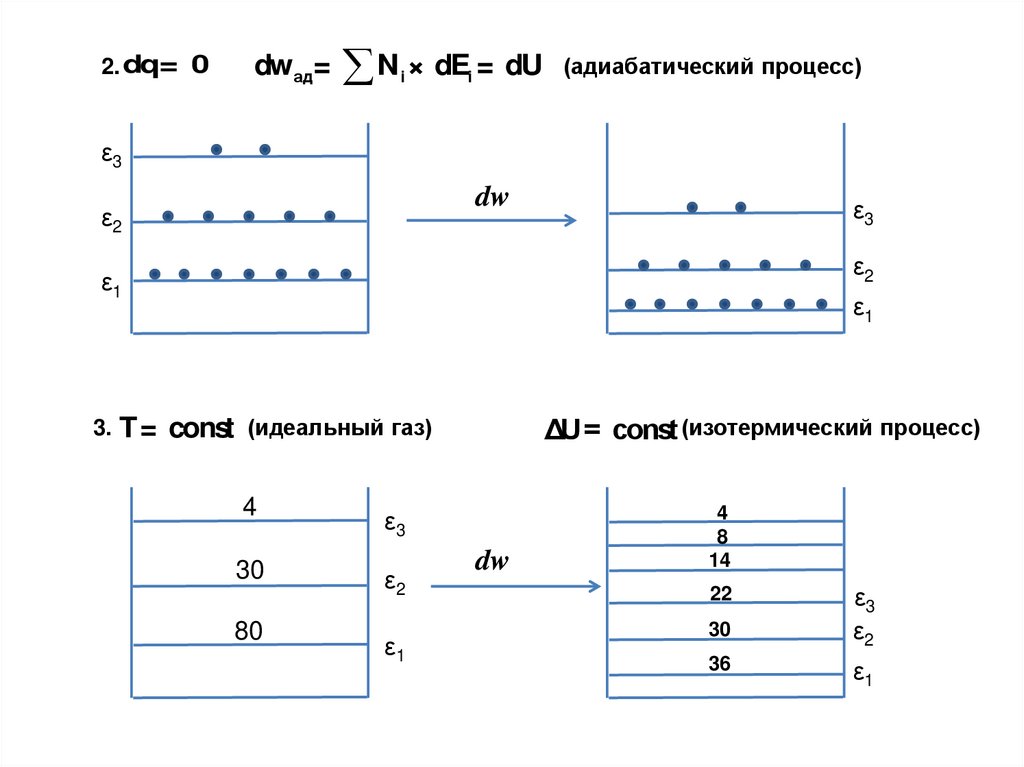
Молекулярная интерпретация теплоты и работы
U – внутренняя энергия системы
U = ΣNi·Ei
Ni – число молекул на i-уровне; Ei – энергия i-уровня
E × dN + N × dE dU= dq+ dw
dq= E × dN = dU (изохорный процесс)
1. dw= 0
dU=
i
i
i
i
i
i
ε3
ε2
ε1
ε3
dq
ε2
ε1
31.
2. dq= 0dwад=
N × dE = dU
i
i
(адиабатический процесс)
ε3
dw
ε2
ε3
ε2
ε1
3.
ε1
ΔU= const (изотермический процесс)
T= const (идеальный газ)
4
ε3
30
ε2
80
ε1
dw
4
8
14
22
30
36
ε3
ε2
ε1
32.
ТермохимияΔ U= q+ w
Δ U= qV
Δ H= qP
q – зависит от пути
qV – не зависит от пути
qP – не зависит от пути
q 0 (экзо)
A+B=C+D
q 0 (эндо)
Δ U= qV
Δ H= qP = Δ U+ p× Δ V
qP - qV = p× Δ V
qP qV (конденсированные среды)
Δ H P0 – стандартная энтальпия реакции (t=25oC, 1 атм)
2Н2(г) + О2(г) → 2Н2О(ж)
H P0 572 кДж
моль
2СН4(г) + 2О2(г) → СО2(г) + 2Н2О(ж) H сж 890 моль
0
кДж
Закон Гесса: Тепловой эффект процесса при P, V и T = const
(1836 г.)
не зависит от промежуточных стадий, а определяется
начальным и конечным состоянием системы
33.
23
1
4
A+B
C+D
6
q12 + q23 + q34 = q16 + q65 + q54
5
Условия выполнения закона Гесса:
1. P и V = const.
2. T = const.
3. Превращение идет до конца.
Задача: С3Н6(г) + 9/2О2(г) → 3СО2(г) + 3Н2О(ж) ΔH P0= ?
С3Н6(г) + Н2(г) → С3Н8(г)
ΔH P0 124 кДж
моль
С3Н8(г) + 5О2(г) → 3СО2(г) + 4Н2О
ΔH P0 2220 кДж
моль
кДж
0
ΔH P 228 моль
Н2О(ж) → Н2(г) + 1/2О2(г)
Σ С3Н6(г) + 9/2О2(г) → 3СО2(г) + 3Н2О(ж)
ΔH P0 124 2220 228 2116 кДж
моль
34.
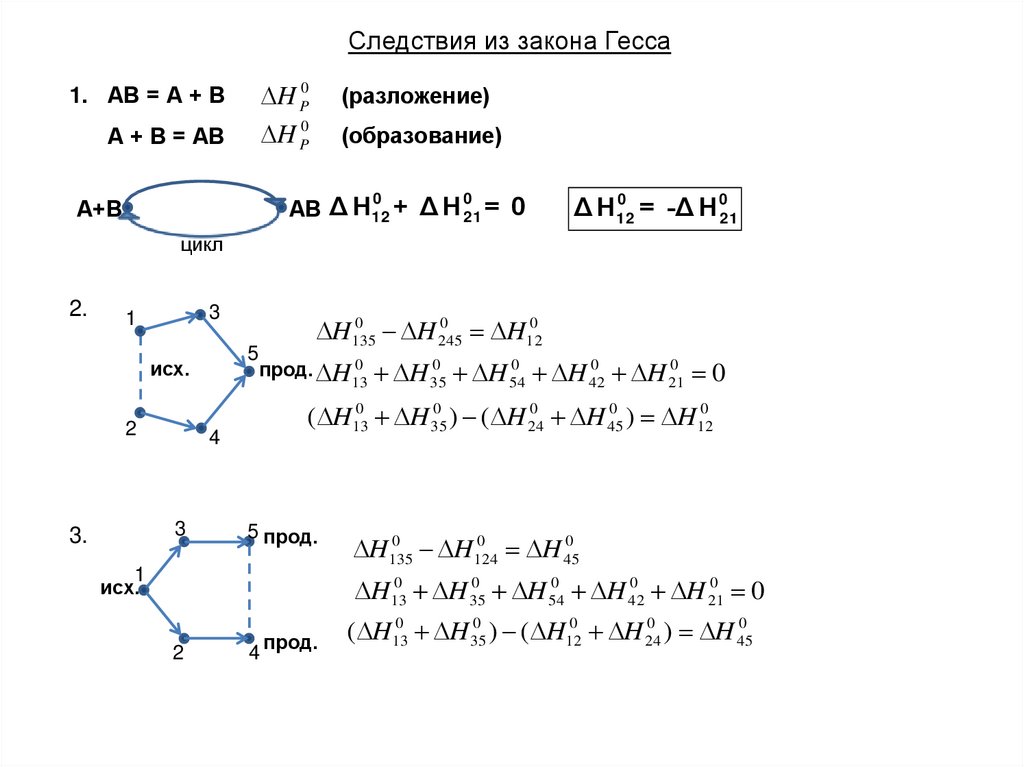
Следствия из закона ГессаH P0
H P0
1. АВ = А + В
А + В = АВ
(разложение)
(образование)
АВ Δ H12 + Δ H 21 = 0
0
А+В
0
0
Δ H12
= -Δ H 021
цикл
2.
3
1
5
исх.
2
прод. H 0
0
0
0
0
H
H
H
H
13
35
54
42
21 0
0
0
0
( H130 H 35
) ( H 24
H 45
) H120
4
3
3.
0
0
H135
H 245
H120
5 прод.
1
исх.
0
0
0
H135
H124
H 45
0
0
0
0
H130 H 35
H 54
H 42
H 21
0
2
4
прод.
0
0
0
( H130 H 35
) ( H120 H 24
) H 45
35.
Стандартная энтальпия образования0
(AB)
А + В = АВ H обр
(25оС, 1 атм)
0
(A) 0
А и В – элементы H обр
0
H обр
(B) 0
0
Н2(г) + 1/2О2(г) → Н2О(ж) H обр
(H2O) 286 моль
кДж
0
С(тв. графит) + 2S(тв. ромб.) → СS2(ж) H обр
(CS2 ) 90 моль
кДж
АВ + CD = АC + ВD H P0 ?
А + В = АВ
0
H обр
(AB)
|АВ = А + В|
0
H обр
(AB)
0
(CD)
C + D = CD H обр
0
А + C = АC H обр
(AC)
0
(BD)
B + D = BD H обр
Δ H 0Р = Δ H
( AC)+ Δ H 0обр( BD) - Δ H 0обр( AB)+ Δ H 0обр( CD)
0
обр
Δ H 0Р=
Δ H
(прод) - Δ H
0
обр
(исх)
0
обр
36.
АВ + CD = АC + ВD Δ H P0 -?АB + O2 = АO + BO
H
CD + O2 = CO + DO H
АC + O2 = АO + CO H
BD + O2 = BO + DO H
Δ H 0Р = Δ H
0
сж
0
сж
0
сж
0
сж
0
сж
( AB)+ Δ H
Δ H 0Р =
Стандартная энтальпия сгорания
(AB)
(CD)
(AC)
(BD)
0
сж
( CD) - Δ H
Δ H
0
сж
0
сж
( AC)+ Δ H
( исх) - Δ H
0
сж
0
сж
( прод)
Уравнение Кирхгофа
Δ H P= f(T)
А+В=C+D
Δ H P = ( HC + HD ) -( H A + HB )
d Δ HP d H C d HD d H A d HB dH i
= CP( i)
=
+
-
+
dT
dT
dT dT
dT
dT
d Δ H P
= CP(C)+ CP(D) - CP(A)+ CP(B)
dT
d Δ HP
= Δ CP – закон Кирхгофа
dT
( BD)
37.
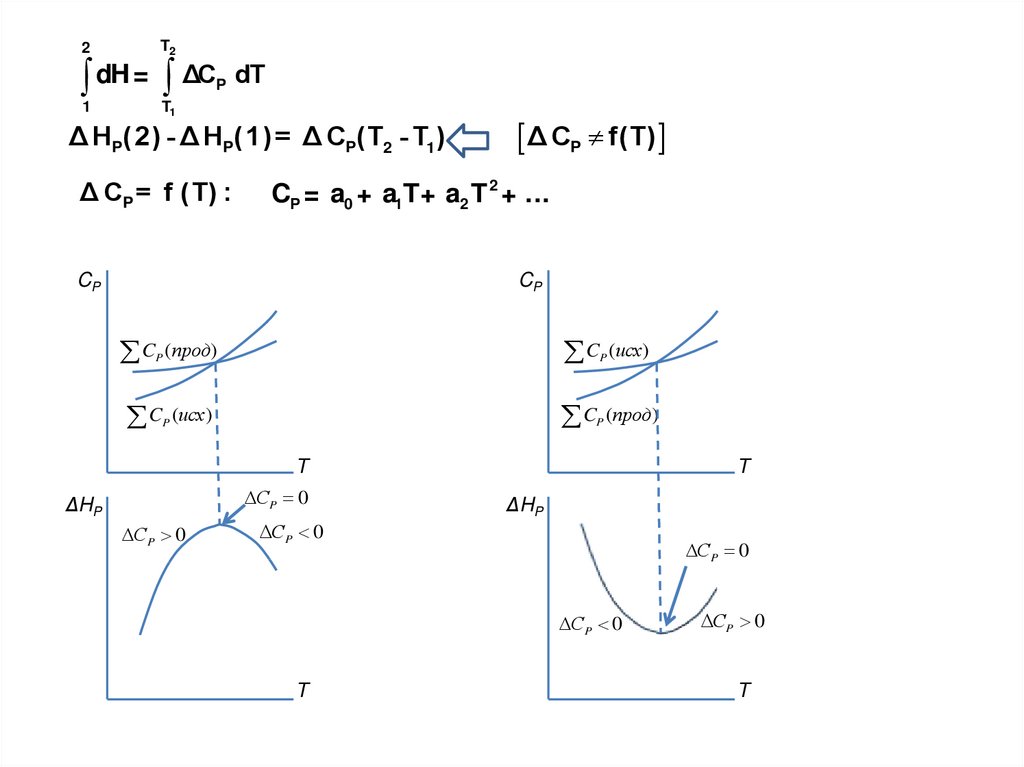
T22
dH = ΔC
P
1
dT
T1
Δ HP(2) - Δ HP(1) = Δ CP(T2 - T1)
Δ CP = f (T) :
Δ CP f(T)
CP = a0 + a1T+ a2 T 2 + ...
CP
CP
C
P
C
(прод)
C
(исх)
C
P
P
P
(исх)
(прод)
T
СP 0
ΔHP
СP 0
СP 0
T
ΔHP
СP 0
СP 0
T
СP 0
T
38.
Термохимическая энтальпия диссоциацииА· + BC → АB + C·
H P0
BC → B· + C· (1) H P0 (1)
0
AB → A· + B· (2) H P (2)
Согласно закону Гесса:
Δ H P0 = Δ H P0 ( 1) - Δ H P0 ( 2)
H P0 (1) – стандартная энтальпия диссоциации ВС [ ДH 0 (BC)]
H P0 (2) – стандартная энтальпия диссоциации АВ [ ДH 0 (AB)]
Δ H P0 = Д H 0 ( BC) - Д H 0 ( AB)
Δ H P0 =
Д H
0
( раз рыв. связ и) - Д H0 ( образ. связ и)
0
Пример: Д H ( CH 3 - H) -?
→ CH · + HBr
Δ H P0 : Br· + CH4 ←
3
Δ H P0 = Д H 0 ( CH 3 - H) - Д H 0 ( HBr)
измеряется
известно
39.
ДH 0 (CH 3 - Cl) ?CH4(г) → CH3·(г) + H·(г)
H P0 ДH 0 (CH3 - H)
0
0
0
ДH 0 (CH3 - H) H обр
(CH3 , г) H обр
(H , г) H обр
(CH4 , г)
известно
вычислим
CH3-Сl(г) → CH3·(г) + Cl·(г)
известно
известно
H P0 ДH 0 (CH3 - Cl)
0
0
0
ДH 0 (CH3 - Сl) H обр
(CH3 , г) H обр
(Cl , г) H обр
(CH3Cl , г)
вычислено
известно
известно
ДH 0 (C - F) ДH 0 (C - Cl) ДH 0 (C - Br ) ДH 0 (C - J)
Ионы в растворе. Энтальпийная шкала ионов.
0
Н+(H2O, 1 М) + ОН-(H2O, 1 М) → H2O(ж) H P 55.835кДж (тепловой эффект нейтрализации)
H2O(ж) → H2(г) + 1/2O2(г)
H P0 285.830кДж
0
H2(г) + 1/2O2(г) → Н+(H2O, 1 М) + ОН-(H2O, 1 М) H P 229.995кДж
0
Принимают H обр (H ,1M) 0кДж
1/2H2(г) + 1/2O2(г) + e- → ОН-(H2O, 1 М)
1/2H2(г) → Н+(H2O, 1 М) + e- H P0 0кДж
0
H обр
(OH ,1M) 229.995кДж
40.
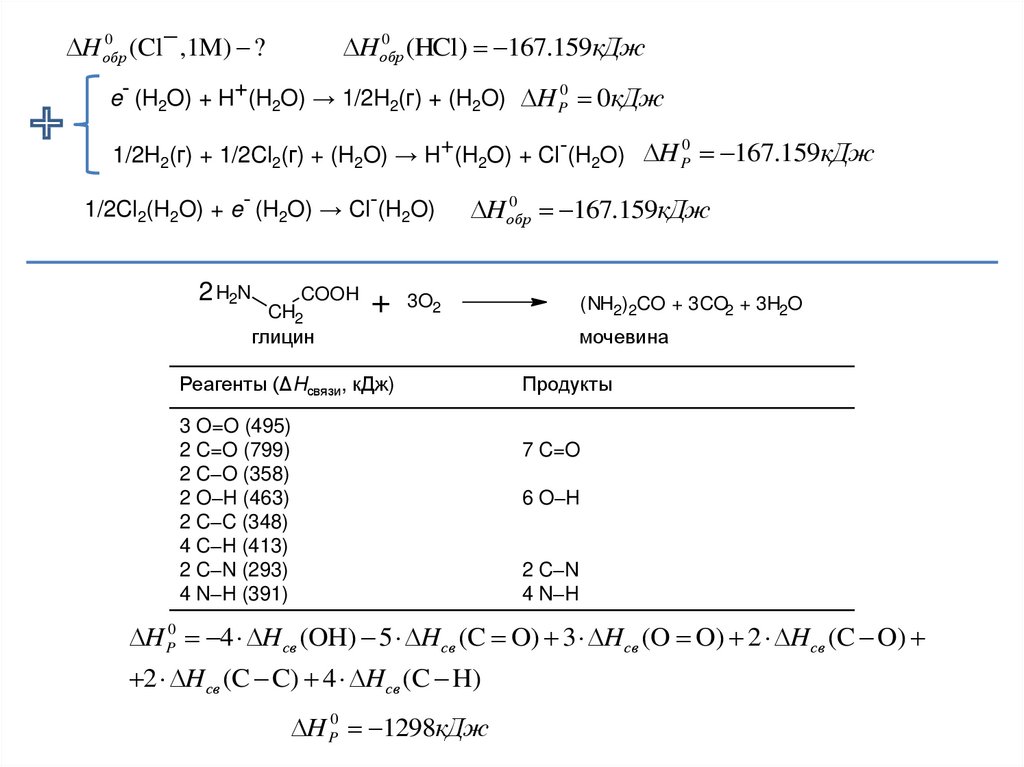
0H обр
(Cl ,1M) ?
0
H обр
(HCl) 167.159кДж
e- (H2O) + H+(H2O) → 1/2H2(г) + (H2O) H P0 0кДж
0
1/2H2(г) + 1/2Cl2(г) + (H2O) → H+(H2O) + Cl-(H2O) H P 167.159кДж
1/2Cl2(H2O) + e- (H2O) → Cl-(H2O)
2 H2N
COOH
CH2
+
0
H обр
167.159кДж
3O2
(NH2)2CO + 3CO2 + 3H2O
глицин
Реагенты (ΔHсвязи, кДж)
3 O=O (495)
2 C=O (799)
2 C–O (358)
2 O–H (463)
2 C–C (348)
4 C–H (413)
2 C–N (293)
4 N–H (391)
мочевина
Продукты
7 C=O
6 O–H
2 C–N
4 N–H
H P0 4 H св (OH) 5 H св (C O) 3 H св (O O) 2 H св (C O)
2 H св (C C) 4 H св (C H)
H P0 1298кДж
41.
II Закон термодинамикиПостулат Клаузиуса: теплота сама не может переходить от холодного тела к горячему.
Клаузиус 1865 г.. Энтропия - «превращение»
dS=
δq
T
Направление самопроизвольного процесса.
Идеальный газ. Энтропия-функция состояния.
dU = 0
p× dV 0
dq= p× dV+ dU
dq 0
dq p× dV dU
=
+
T
T
T
p R
=
T V
Процесс циклический и обратимый
dU
=
T
dqобр
R
- dV
T
V
dU
=
T
cV dT
=
T
c
V
dlnT = 0
dU= cV dT
42.
RV dV
Rd ln V 0
dqобр
= 0
T
Необратимые процессы (w – работа)
dw
dw
T T обр dw ( dw)обр
dqобр
0
dw
T
0
T обр
dU
0
T 0
dq
dU
dw
dw
0
T T T
T
2
1
dq
0 сумма приведенных
T
теплот ≤ 0
Если в процессе хотя бы одна стадия необратима,
то весь процесс необратим
P
необрат.
0
обрат.
2
2
V
1
2
2
dqобр
dqобр
dq
dq
dq
T 1 T 2 T
T
T
1
1
1
dq
S 0
T
Изолированная система
S
dq
– неравенство Клаузиуса
T
dq=0 Δ S 0
43.
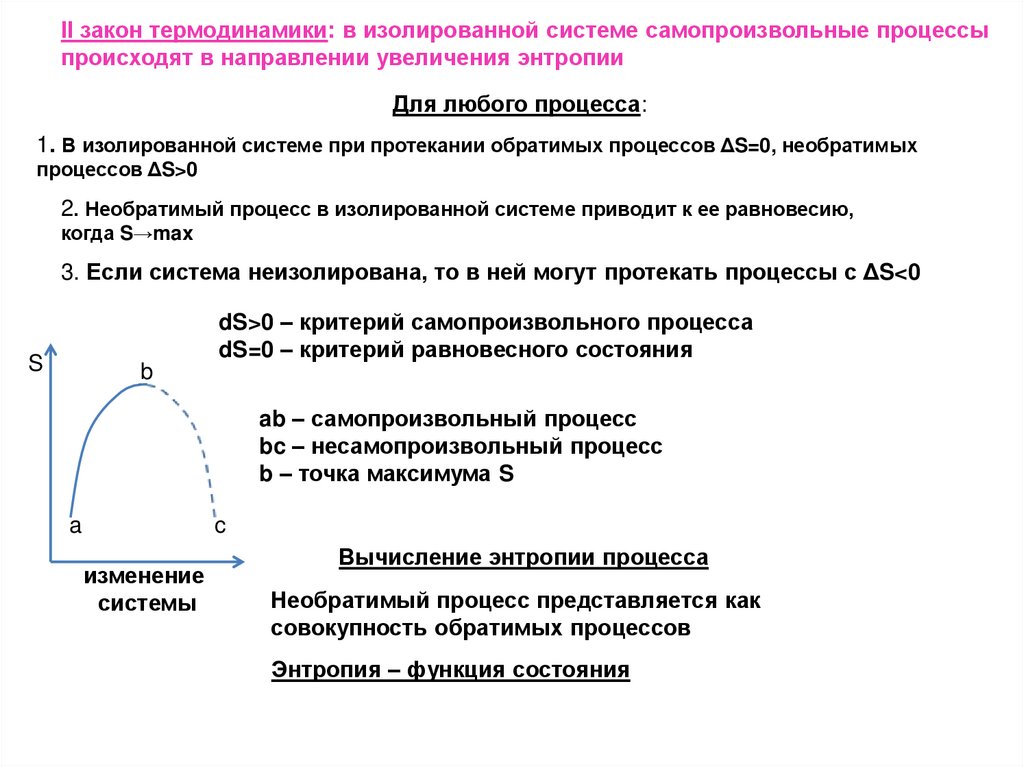
II закон термодинамики: в изолированной системе самопроизвольные процессыпроисходят в направлении увеличения энтропии
Для любого процесса:
1. В изолированной системе при протекании обратимых процессов ΔS=0, необратимых
процессов ΔS>0
2. Необратимый процесс в изолированной системе приводит к ее равновесию,
когда S→max
3. Если система неизолирована, то в ней могут протекать процессы с ΔS<0
dS>0 – критерий самопроизвольного процесса
dS=0 – критерий равновесного состояния
S
b
ab – самопроизвольный процесс
bc – несамопроизвольный процесс
b – точка максимума S
a
c
изменение
системы
Вычисление энтропии процесса
Необратимый процесс представляется как
совокупность обратимых процессов
Энтропия – функция состояния
44.
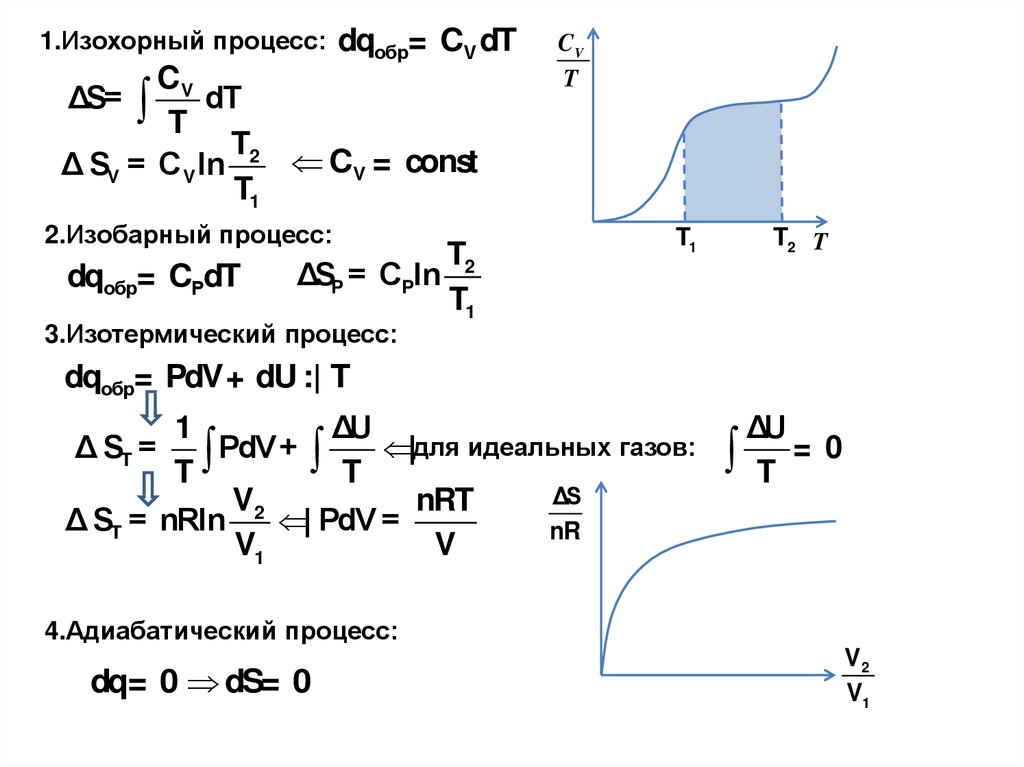
1.Изохорный процесс:dqобр= CV dT
CV
ΔS=
dT
T
T
Δ SV = C V ln 2 CV = const
T1
2.Изобарный процесс:
dqобр= CPdT
T2
ΔSP = CPln
T1
CV
T
T1
T2 T
3.Изотермический процесс:
dqобр= PdV+ dU :| T
1
ΔU
PdV+
|для идеальных газов:
T
T
ΔS
V2
nRT
Δ ST = nRln
| PdV =
nR
V1
V
Δ ST =
ΔU
T=0
4.Адиабатический процесс:
dq= 0 dS= 0
V2
V1
45.
EE
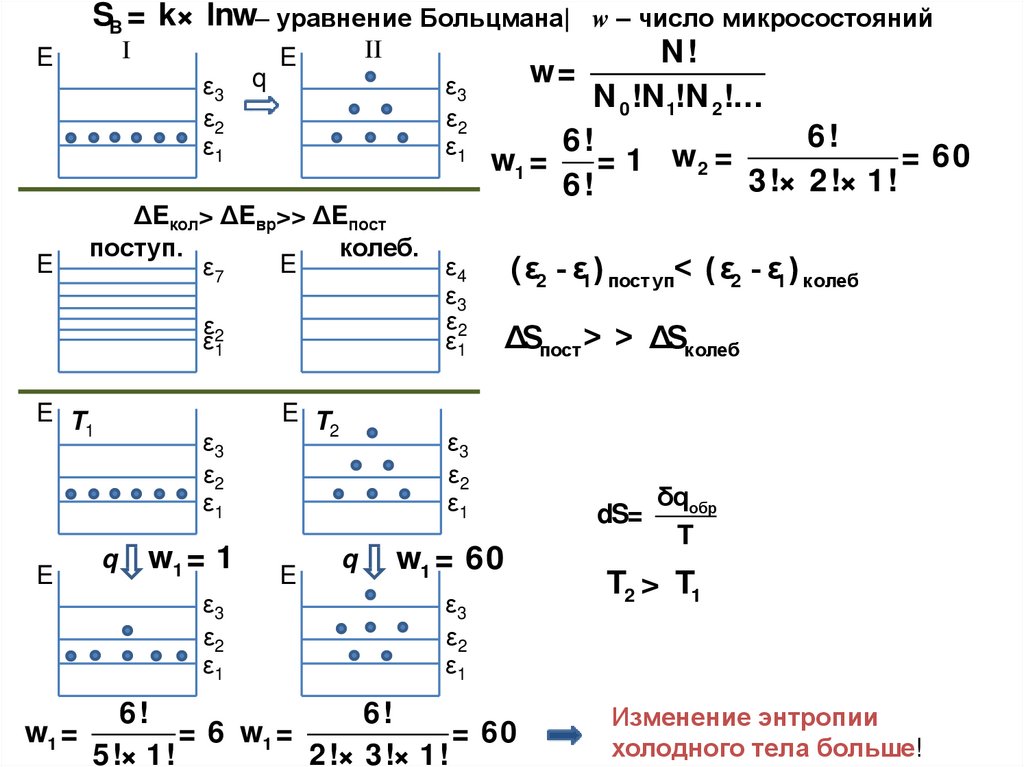
SB = k× lnw– уравнение Больцмана| w – число микросостояний
II
I
N!
E
w=
ε3 q
ε3
N 0 !N 1!N 2!...
ε2
ε2
6!
6!
ε1
ε1
= 60
w1 =
= 1 w2 =
3!× 2!× 1!
6!
ΔEкол> ΔEвр>> ΔEпост
поступ.
колеб.
E
ε4
ε7
ε3
ε2
ε
ε12
ε1
E T
1
E
ε3
ε2
ε1
q
w1 = 1
ε3
ε2
ε1
E T
2
E
(ε2 - ε1 ) пост уп< (ε2 - ε1 ) к олеб
ΔSпост > > ΔSк олеб
ε3
ε2
ε1
q
w1 = 60
ε3
ε2
ε1
6!
6!
w1 =
= 6 w1 =
= 60
5!× 1!
2!× 3!× 1!
dS=
δqобр
T
T2 > T1
Изменение энтропии
холодного тела больше!
46.
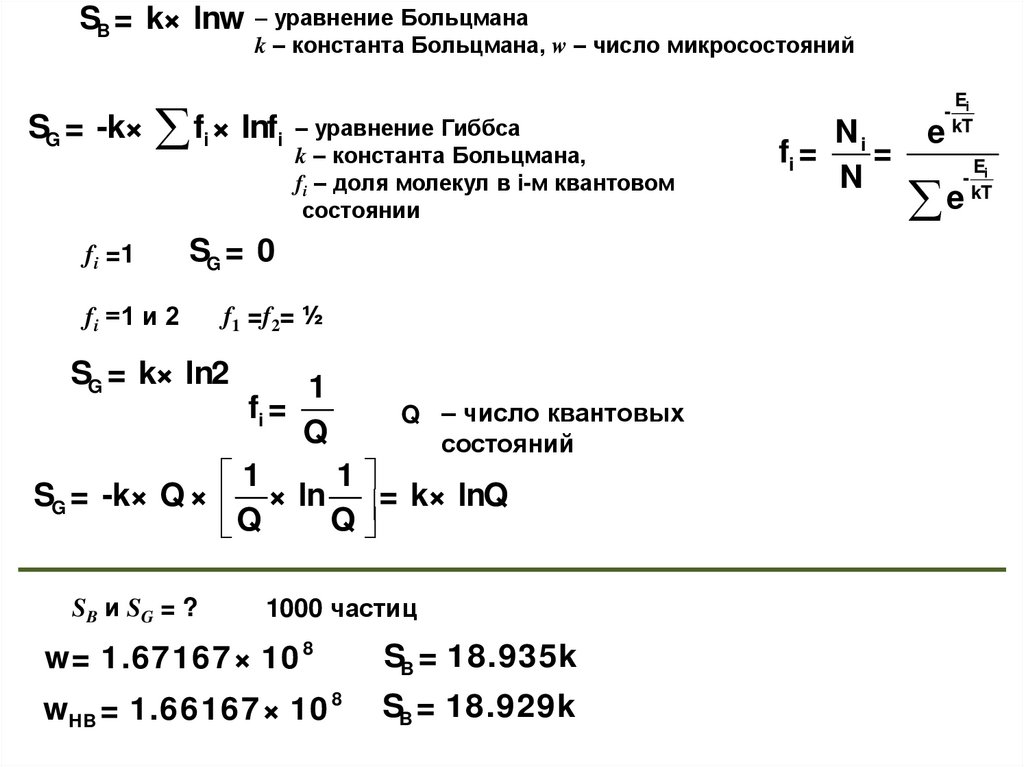
SB = k× lnwSG = -k×
– уравнение Больцмана
k – константа Больцмана, w – число микросостояний
f × lnf
fi =1
i
i
– уравнение Гиббса
k – константа Больцмана,
fi – доля молекул в i-м квантовом
состоянии
SG = 0
fi =1 и 2
f1 =f2= ½
SG = k× ln2
fi =
1
Q
Q – число квантовых
состояний
1
1
SG = -k× Q × × ln = k× lnQ
Q
Q
SB и SG = ?
1000 частиц
w= 1.67167× 10 8
SB = 18.935k
wHB = 1.66167× 10 8
SB = 18.929k
fi =
Ni
=
N
-
e
Ei
kT
e
-
Ei
kT
47.
f0 = 0.997SG = ?
SG = -k×
f1 = 0.003
f × lnf = -k× 0.997× ln0.997 + 0.003× ln0.00 3 =
i
i
= 0.020423k
(на 1 частицу)
На 1000 частиц
SG = 20.423k
Энтропия в химической термодинамике
1. Образование двухатомной молекулы: ΔS<0
2. Испарение и сублимация: ΔS>0
3. Изохорный процесс: ΔS>0
4. Адиабатический процесс (обратимый): ΔS=0
5. Изотермический процесс: ΔS>0
48.
Теория термодинамического равновесияdq= T× dS
dq= dU+ P× dV
dU= T× dS- P× dV
– обратимые изменения
Уравнение Клаузиуса
dU S, V = 0 (при равновесии)
H= U+ PV
dH= T× dS- P× dV+ V× dP+ P× dV
dH= T× dS+ V× dP– обратимые изменения dH S,P = 0 (при равновесии)
A = U - T× S– функция Гельмгольца
dA= dU - T× dS- S× dT= T× dS- P× dV - T× dS- S× dT
dA= -S× dT - P× dV– обратимые изменения dA T, V = 0 (при равновесии)
A – функция максимальной работы
dA = dU - T×dS =
T= const;
S× dT= 0
= dqобр - T×dS + dw обр
dqобр= T× dS
dA = dw обр
49.
G= H - T× S– функция ГиббсаdG= dH - T× dS- S× dT= T× dS+ V× dP- T× dS- S× dT
dG= -S× dT+ V× dP– обратимые изменения dGT,P = 0 (при равновесии)
G – функция полезной работы
Работа – процесс преодоления системой сил внешнего воздействия
Силы: 1) давление; 2) поверхностное натяжение; 3) электрические; и т.д.
dw= -P× dV+ x× dy+ z× dQ+ m× dN+ ...
P, x, z, m- интенсивные свойства
dV, dy, dQ, dN - приращения экстенсивных свойств
| dqобр= T× dS
T= const; dG= dqобр+ dw обр+ d(PV) - T× dS=
= dqобр+ dw' - P× dV+ P× dV+ V× dP- T× dS| dw обр= dw' - P× dV
dG= dw'+ V× dP | P= const|
dG= dw' (при P и T = const)
50.
Почему G и A?N= U+ T× S, dN= dU+ TdS+ SdT= 2TdS+ SdT -PdV
M = U+ P× T (P×T – не имеет размерность энергии)
Полный дифференциал
f = f(x, y)
P~ f(V, T) (PV=RT)
f
f
df = dx + dy = m dx +n dy
x y
y x
f
f
m n
= (Эйлер)
y x y x x y x y
y x x y
функция состояния
Пример: 1. dT для PV=RT?
2. T – функция состояния?
3. Значение Т не зависит от пути?
PV
T
T
dV
+
dP
T
=
V
P
R
P
V
1. dT =
P
T
=
;
V P R
V
T
=
P
V R
dT =
P
V
dV + dP
R
R
51.
PR =
2.
P
V
V
R = 1 T = PV
V
R
R
P
dT =
P
V
dV + dP
R
R
dT – полный дифференциал, T – функция состояния
3. P
PB, VB
PB, VA
TA - TB = const (I или II)
I путь
II
PA, VA
=
V
a
a
a
P
a R dV+
P
V
Va - Vb + Pa - Pb
R
R
b
P
V
dV +
R
a
P
b R dV+
P
V
V
V
+
a b
Pa - Pb
R
R
II путь T - T = dT = P dV+
A
B
b
b R
=
b
P
TA - TB = dT = dV+
R
b
b
I
PA, VB
a
PV = RT dT =
a
V
b R dP+
0
dP
R
a
V
a R dP=
0
a
V
b R dP+
V
a R dP=
0
0
52.
Фундаментальные уравнения термодинамикиT
1. Внутренняя энергия U
dU= T× dS- P× dV
U= f(S, V)
f
=
y x y x
x y
–P
f
x y x y
U
U
dU=
× dS+
× dV
x→S, y→V
S V
V S
U
U
T
P
= - – уравнение Максвелла
= -P
= T
V S
S V
V S
S V
2. Энтальпия H
H= U+ PV
H= f(S,P)
dH= dU+ P× dV+ V× dP= T× dS+ V× dP
H
H
dH=
×
dS+
× dP
S P
P S
H
= T
S P
H
= V
P S
T
=
P S
T
x y
f
=
y x y x
V
f
x y x y
V
– уравнение Максвелла
S P
53.
3. Энергия Гельмгольца AA = U - TS
dA= dU - T× dS-S× dT= T× dS-P× dV - T× dS-S× dT= -P× dV -S× dT
A
A
A= f(V, T)
dA =
×
dT+
× dV
–P
–S
T V
V T
x y
A
A
f
f
= -S
= -P
=
T V
V T
y x y x x y x y
P
S
=
– уравнение Максвелла
T V V T
4. Энергия Гиббса G
G= H - TS
dG= dH - T× dS-S× dT= T× dS+ V× dP- T× dS-S× dT= V× dP-S× dT
G
G
dG=
× dT+
× dP G= f(P, T)
V
–S
T P
P T
x y
f
G
f
G
=
-S
=
V
=
T
P
P
T
y
x
y x
x y x y
S
V
=
– уравнение Максвелла
T P
P T
54.
Фундаментальные уравнения равновесной термодинамикиУравнение
Комментарии
Комбинация I и II законов (Клаузиус)
dU= T× dS- P× dV
H= U+ PV
A = U - TS
G= H - TS
Базовые определения
dH= T× dS+ V× dP
dA= -S× dT - P× dV
dG= -S× dT+ V× dP
T
=
V S
P
-
S V
T
=
P S
V
S P
Альтернативные определения
комбинации I и II законов
S
V =
T
P
T
V
S
P =
T
V
-
T P
Уравнения Максвелла
55.
Для однокомпонентной системы: U, H, A, G, S, P, V и T336 – частных производных для однокомпонентных систем
1. Циклическое правило
x y Z
y × Z × x = -1
x
y
Z
Z= Z(x, y)
Задача
RT P
R
=
V
T V V
PV T
P
( y) T =
=
R
V P R
RT V
RT
(z ) V =
=
P P T
P2
(x ) P=
PV = RT
2. Правило деления
Z= Z(x, y)
Z
=
t
x
Z
Z
dz =
dx
+
dy
x y
y x
Z
×
x
y
x
+
t x
Z
y ×
x
0
|×
1
( t)x
Z
y
=
×
t
y
x
x
y
t x
56.
1S
H
CP =
=
T×
|
dH=
T×
dS+
V×
dP
|
×
T
( T) P
T P
P
Задача CP - CV = ?
S
U
CV =
=
T×
| dU= T× dS- P× dV
T V
T V
S= f(T, V)
1
S
S
dS=
dT+
dV
|
×
V
( T) P
T V
T
S S
S S V
T S V
=
×
+
×
+
×
T T
P V T P V T T P T V V T T P
1
S
S V
S
S V
CP - CV = T×
+
T×
×
T×
=
T×
×
=
T V
V T T P
T V
V T T P
P
V
Уравнение Максвелла
= T×
×
Циклическое
правило
T V T P
S
P
V
×
T P
T
P
×
= -1
P V V T
P
-
=
T V
V P
×
T P V T
V = T
V
T
57.
2P V
CP - CV = -T×
×
V T T P
1
P
=
; k – коэффициент изотермической сжимаемости
k× V
V T
V
= α × V; α – коэффициент термического расширения
T P
α2
CP - CV = T× V×
k
Как определить CP при различных давлениях?
S
CP
H
C
=
=
T×
=
?
P
T
P
T
P
P
T
S
CP
=
T×
= T×
P
T
P T P T
S
CP
P = T× T P
T
T P
ΔH= T× dS+ V× dP
S
T P T P
S
P =
T
V
-
T P
V
CP
2
=
-T
×
=
-T
×
V
×
α
T T
P P
P T
f
=
y x y x
f x=T
y=P
x y x y f=S
(Эйлер)
Ур-ние Максвелла
58.
CP (P) = - T × V × α 2 × dp Если известно, что α = f(P) , то СP при любом PКак определить CV при разных объемах?
CV
S
U
=
?
C
=
=
T×
V
T
V
T
V
V
T
S
CV
=
T×
= T×
V
T
V T V T
S
CV
V = T× T V
T V
T
S
T
V
T V
dU= T× dS- P× dV| ×
= T×
α
-P
k
f
=
y x y x
f x=T
y=V
x y x y f=S
(Эйлер)
S
P
=
V
T T V
Ур-ние Максвелла
2P
CV
V = T× T 2
T
Внутреннее давление
dU= T× dS- P× dV
U
= 0
V T
2P
CV (V) = T× 2 × dV
T
(идеальный газ)
1
S
U
V
=
T×
P×
V
( V) T V T
V T
T
S
P
=
V
T T V
ур-ние Максвелла
S
V =
T
P
=
T V
S
U
=
T×
- P=
V T
V T
P
-
×
V T
α
V
=
T P k
Циклическое правило
59.
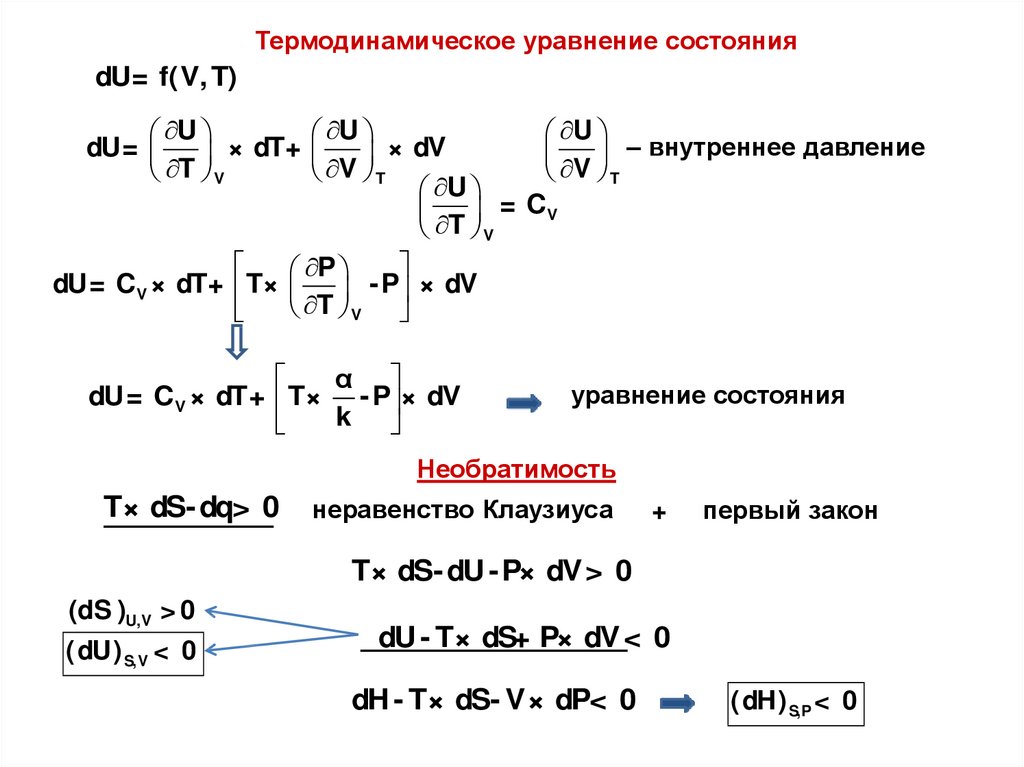
Термодинамическое уравнение состоянияdU= f(V, T)
U
U
U
dU=
×
dT+
×
dV
– внутреннее давление
T V
V T
V T
U
= CV
T V
P
dU= CV × dT+ T×
P
× dV
T V
α
dU= CV × dT+ T× - P × dV
k
уравнение состояния
Необратимость
T× dS- dq> 0
неравенство Клаузиуса
+
первый закон
T× dS- dU - P× dV > 0
(dS )U,V > 0
( dU) S, V < 0
dU - T× dS+ P× dV < 0
dH - T× dS- V× dP< 0
( dH) S,P < 0
60.
dA+ T× dS+ S× dT - T× dS+ P× dV < 0A=
U - TS
dA+ S× dT+ P× dV < 0
( dA) T, V < 0
dG= dH - T× dS- S× dT=
G= H - TS
= dU+ P× dV+ V× dP- T× dS- S× dT
dG- P× dV+ T× dS+ V× dP- T× dS- S× dT+ P× dV < 0
dG+ V× dP- S× dT< 0
( dG) T,P < 0
61.
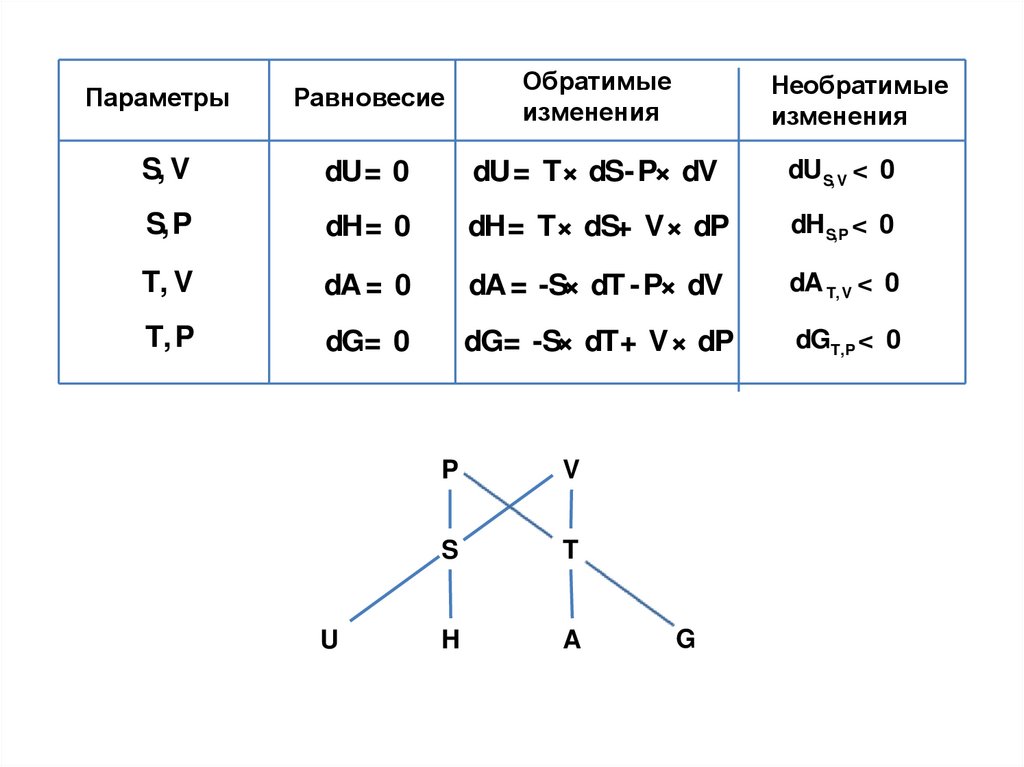
РавновесиеОбратимые
изменения
S, V
dU= 0
dU= T× dS- P× dV
dUS, V < 0
S,P
dH= 0
dH= T× dS+ V× dP
dH S,P < 0
T, V
dA = 0
dA= -S× dT - P× dV
dA T, V < 0
T, P
dG= 0
dG= -S× dT+ V× dP
dGT,P < 0
Параметры
U
P
V
S
T
H
A
Необратимые
изменения
G
62.
Фазовые равновесия в чистых веществахP
ж.
Фазовая диаграмма
Т – тройная точка
тв.
Т
газ
T
Фаза – совокупность частей
гетерогенной системы,
разделенных поверхностью
раздела и
характеризующихся
одинаковыми свойствами
газ
H2O(ж)→H2O(г)
CO2(тв)→CO2(г)
ж
P(ж)=P(газ)=P(атм) → условие кипения
Работа:
W= - P× dV = -P× ΔV P= const
W= -P× V(г)
W= -n× RT
ΔV=V(г) – V(ж. или тв.) ≈ V(г)
(для идеального газа)
ΔU= ΔH - P× ΔV = qP - P× ΔV
dqP qP ΔH
Энтропия: ΔS=
T = T = T | P, T = const
Энергия:
Энергия Гельмгольца:Δ A=
Δ U - T× Δ S
| Δ H= T× Δ S
63.
Энергия Гиббса:G= H - T× S
ΔG= ΔH - T× ΔS-S× ΔT= ΔH - T× ΔS| T,P= const
dq
q
ΔH
ΔG= 0| ΔH= T× ΔS| | ΔS= P = P =
T
T
T
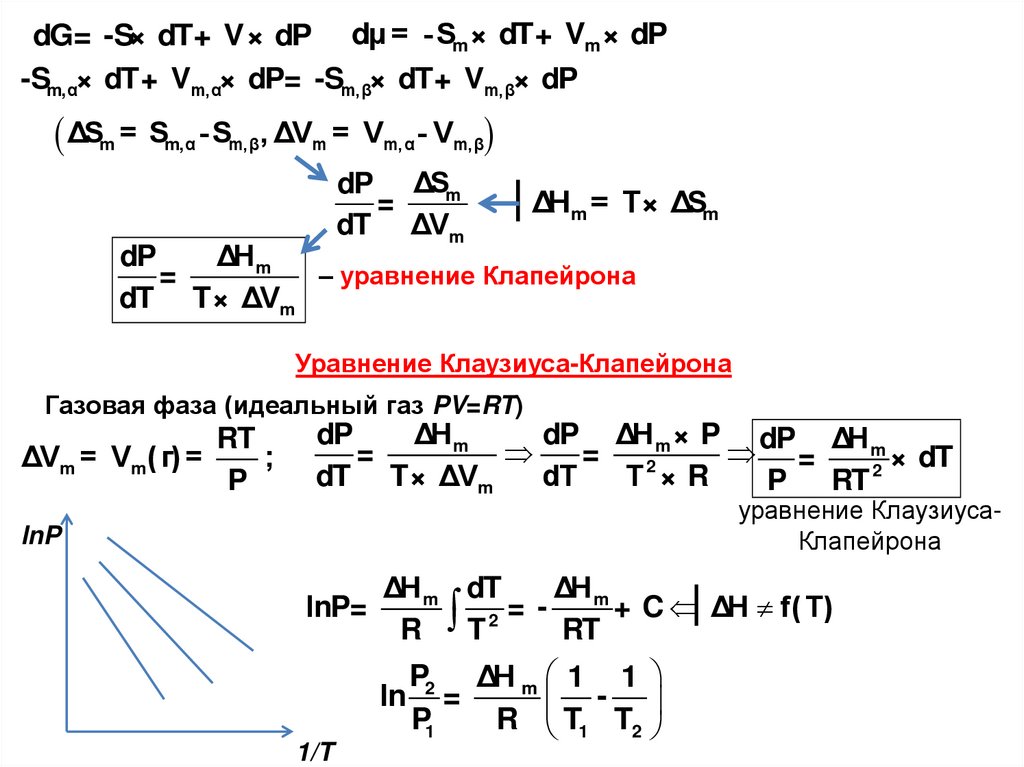
Уравнение Клапейрона
P= f(T) -? (α и β – фазы)
G
μ α = α – химпотенциал однокомпонентной системы в фазе α
nα
Gβ
– химпотенциал однокомпонентной системы в фазе β
μ β=
nβ
G= nα× μ α+ n β× μ β; nα, nβ – число молей в α и β
ΔG= 0 μ α× dn α+ μβ× dn β= 0
P
α фаза
dnα= -dnβ μ α= μβ – условие равновесия
2
μ' α= μ' β
dμ α = dμ β
μ α= μ β
1
β фаза
T
μ 'α = μ α +dμ α
μ 'β = μ β +dμ β
μ' α= μ' β
μα+ dμα= μ β + dμ β
dμ α = dμβ
64.
dG= -S× dT+ V× dP dμ = - Sm × dT+ Vm × dP-Sm,α× dT+ Vm,α× dP= -Sm,β× dT+ Vm,β× dP
ΔS
m
= Sm,α - Sm,β, ΔVm = Vm,α - Vm,β
dP ΔSm
=
dT ΔVm
| ΔH
m
= T× ΔSm
ΔH m
dP
– уравнение Клапейрона
=
dT T× ΔVm
Уравнение Клаузиуса-Клапейрона
Газовая фаза (идеальный газ PV=RT)
RT
ΔVm = Vm ( г) =
;
P
ΔH m
dP
dP ΔH m × P dP ΔH m
=
=
=
× dT
2
2
dT T× ΔVm
dT
T ×R
P RT
уравнение КлаузиусаКлапейрона
lnP
lnP=
ΔH m dT
ΔH m
=
+ C | ΔH f( T)
2
R
T
RT
ln
1/T
P2 ΔH m 1 1
=
-
P1
R T1 T2
65.
ΔH m = f(T)d ΔH m
2
= ΔCp ; Cp = a+ b× T+ c× T + ...
dT
Δb
ΔH m = ΔH 0 + Δa T +
× T 2 + ... ΔH 0 f( T)
2
B
logP= A+ + C× logT (уравнение Рэнкина)
T
B
(уравнение Антуана)
logP= A+
T+ C
ΔHm – энтальпия фазового перехода; ΔHисп – (ж-газ), ΔHпл – (тв-ж), ΔHсубл – (тв-газ)
dG= -S× dT+ V× dP;
μ i= f(T, P)
μi
тв
Sm (г ) > Sm (ж) > Sm (т в)
ж
μi
= -Sm
T P
газ
T
μi
μi
= Vm
P T
газ
тв
ж
Vm(г ) > Vm(ж) > Vm(т в)
P
66.
fHm
f
Hm
Um
Um
Vm
Vm
Sm
Sm
T
T
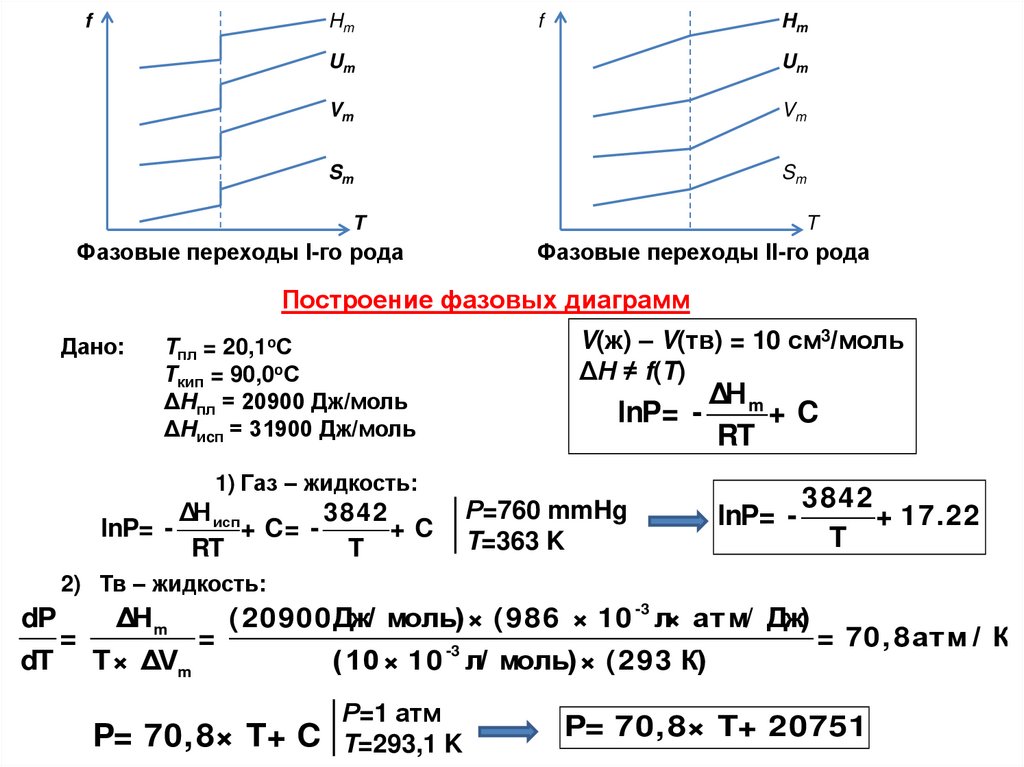
Фазовые переходы I-го рода
Фазовые переходы II-го рода
Построение фазовых диаграмм
Дано:
V(ж) – V(тв) = 10 см3/моль
ΔН ≠ f(T)
Tпл = 20,1оС
Ткип = 90,0оС
ΔНпл = 20900 Дж/моль
ΔНиcп = 31900 Дж/моль
lnP= -
1) Газ – жидкость:
Р=760 mmHg
T=363 K
ΔH исп
3842
lnP= + C= +C
RT
T
ΔH m
+C
RT
lnP= -
3842
+ 17.22
T
2) Тв – жидкость:
dP
dT
=
ΔH m
T× ΔVm
=
( 20900Дж/ моль)× ( 986 × 10 -3 л× а т м/ Дж)
P= 70,8× T+ C
( 10 × 10 л/ моль)× ( 293 К)
-3
Р=1 атм
T=293,1 K
= 70, 8а т м / К
P= 70,8× T+ 20751
67.
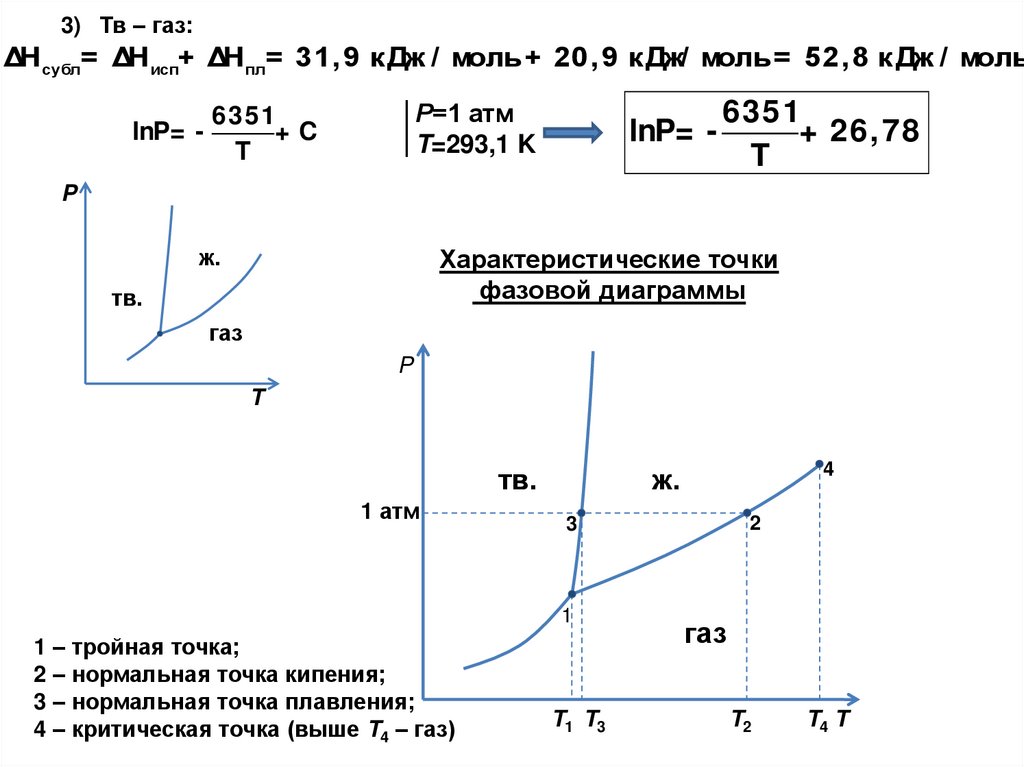
3) Тв – газ:ΔH субл= ΔH исп+ ΔH пл= 31, 9 к Дж / моль + 20, 9 к Дж/ моль = 52, 8 к Дж / моль
lnP= -
Р=1 атм
T=293,1 K
6351
+C
T
6351
lnP= + 26, 78
T
P
ж.
Характеристические точки
фазовой диаграммы
тв.
газ
P
T
тв.
1 атм
2
3
1
1 – тройная точка;
2 – нормальная точка кипения;
3 – нормальная точка плавления;
4 – критическая точка (выше T4 – газ)
4
ж.
T1 T3
газ
T2
T4 T
68.
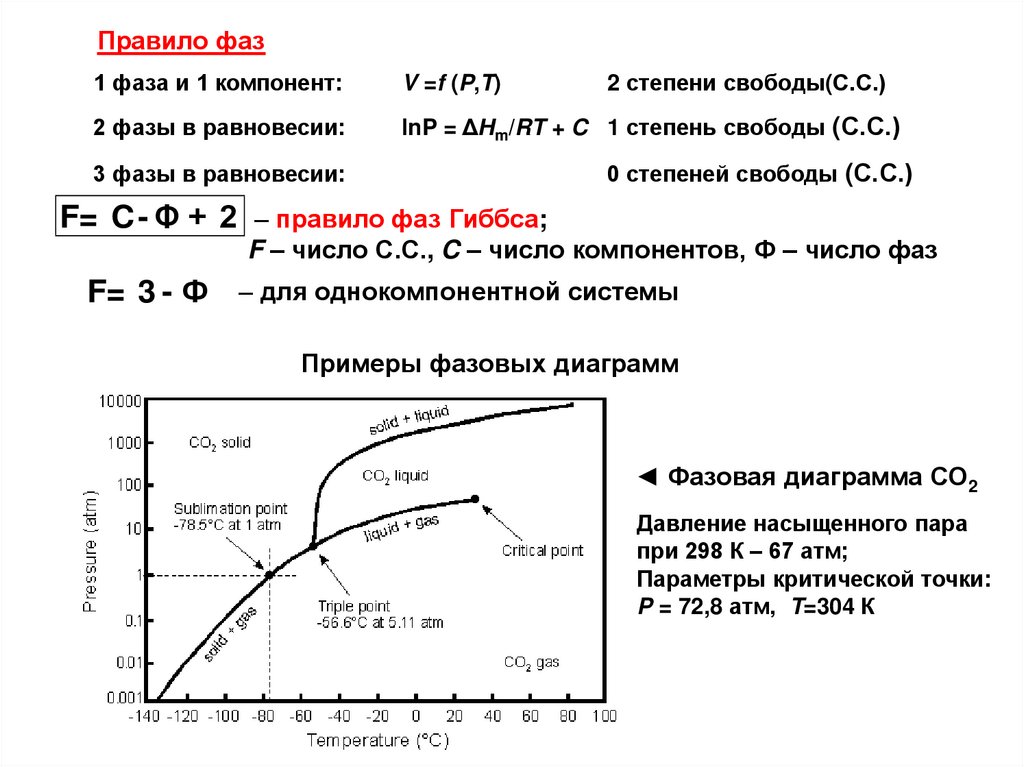
Правило фаз1 фаза и 1 компонент:
V =f (P,T)
2 фазы в равновесии:
lnP = ΔНm/RT + C 1 степень свободы (С.С.)
3 фазы в равновесии:
2 степени свободы(С.С.)
0 степеней свободы (С.С.)
F= C - Φ + 2 – правило фаз Гиббса;
F – число С.С., C – число компонентов, Ф – число фаз
F= 3 - Ф – для однокомпонентной системы
Примеры фазовых диаграмм
◄ Фазовая диаграмма СО2
Давление насыщенного пара
при 298 К – 67 атм;
Параметры критической точки:
P = 72,8 атм, T=304 К
69.
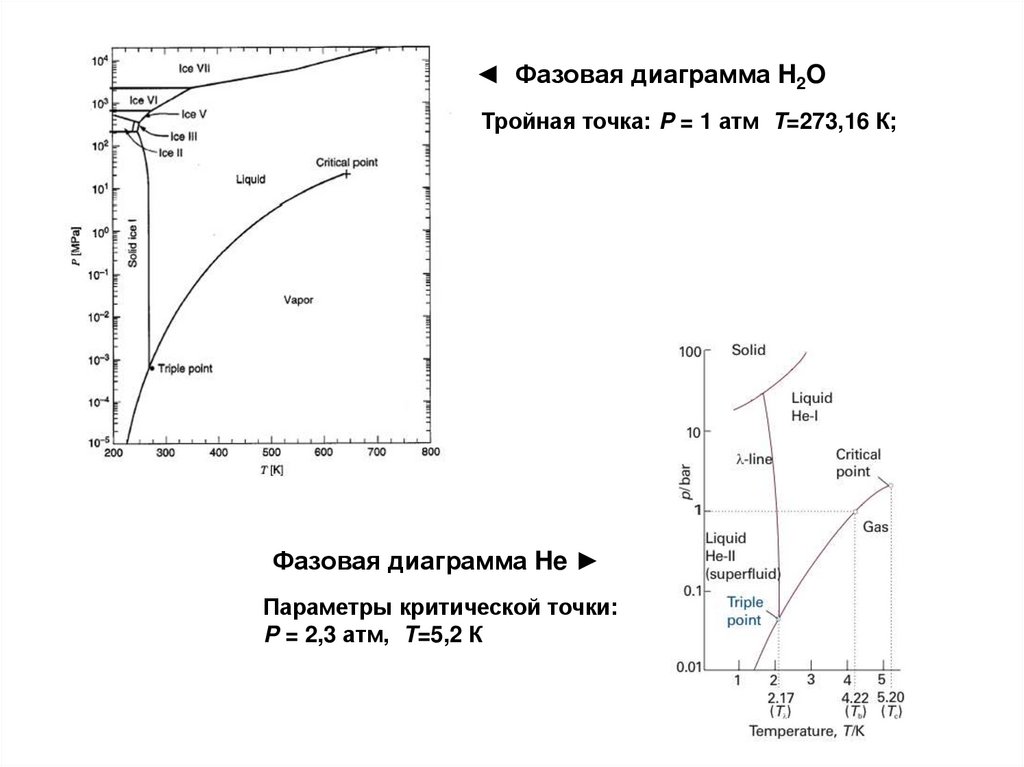
◄ Фазовая диаграмма H2OТройная точка: P = 1 атм T=273,16 К;
Фазовая диаграмма He
Параметры критической точки:
P = 2,3 атм, T=5,2 К
70.
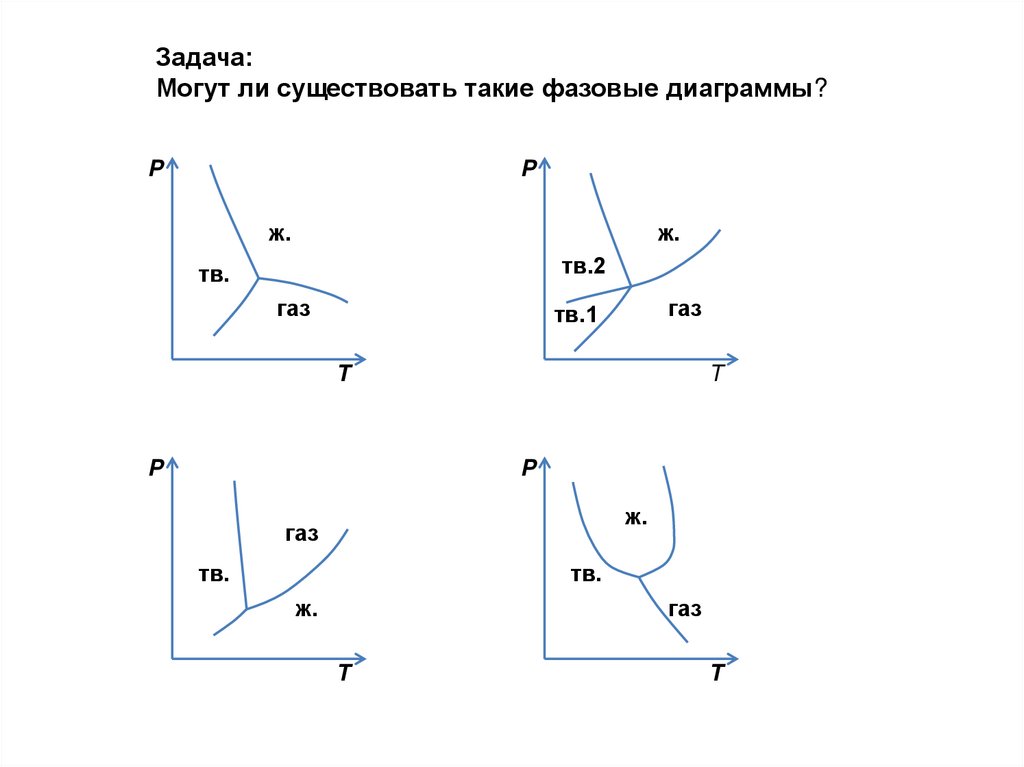
Задача:Могут ли существовать такие фазовые диаграммы?
P
P
ж.
ж.
тв.2
тв.
газ
газ
тв.1
T
P
T
P
ж.
газ
тв.
тв.
ж.
газ
T
T
71.
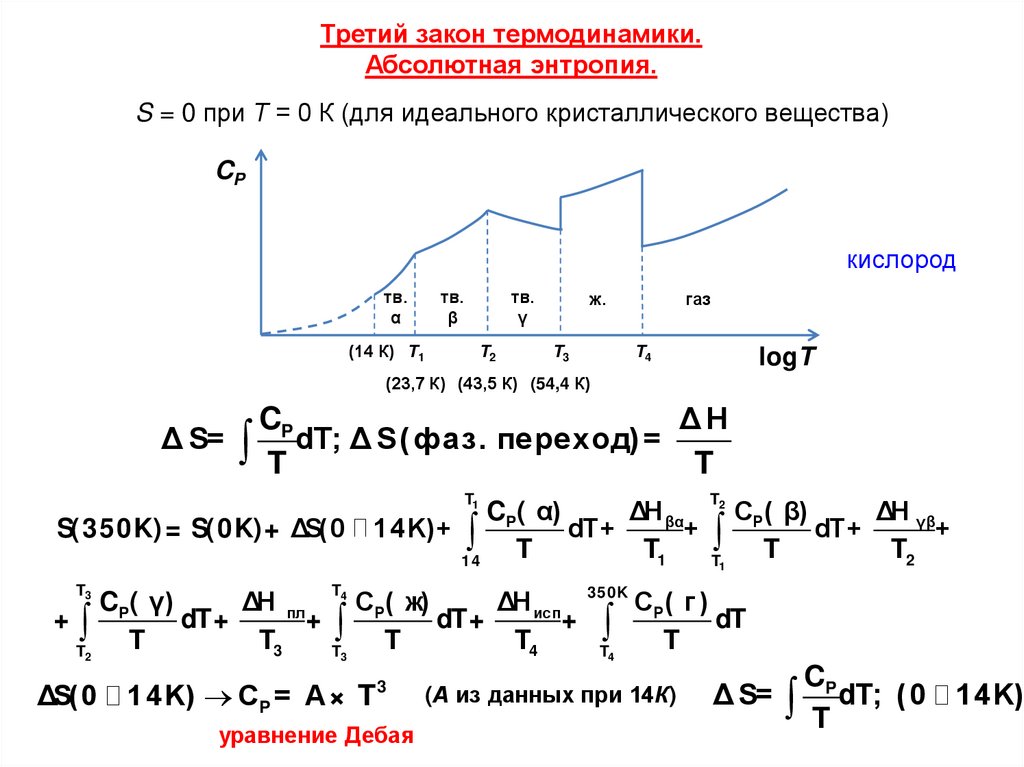
Третий закон термодинамики.Абсолютная энтропия.
S = 0 при Т = 0 К (для идеального кристаллического вещества)
CP
кислород
тв.
α
тв.
β
тв.
γ
(14 К) T1
T2
газ
ж.
T3
T4
logT
(23,7 К) (43,5 К) (54,4 К)
Δ S=
ΔH
CP
Δ
S
(
ф
а
з
.
п
е
р
е
х
о
д
)
=
dT;
T
T
T1
S( 350K) = S( 0K)+ ΔS( 0
T3
+
T2
ΔS( 0
CP ( γ)
ΔH пл
dT+
+
T
T3
T4
T3
C ( α)
ΔH βα
14K)+ P
dT+
+
T
T1
14
CP ( ж)
ΔH исп
dT+
+
T
T4
14K) CP = A× T 3
уравнение Дебая
350K
T4
T2
T1
CP ( β)
ΔH γβ
dT+
+
T
T2
CP ( г )
dT
T
(А из данных при 14К)
Δ S=
CP
T dT; ( 0
14K)
72.
Общая теория переменного составаКонцентрационные шкалы:
m
n=
M
Моль (n)
m – навеска в г
M – молекулярный вес
n1
n1 + n2
n
Молярность (C)
C=
V – объем р-ра
V
n1
× Pt = Х1× Pt Pt – общее давление
Парциальное давление (Pi) P1 =
n1 + n2
Мольная доля (xi)
Х1 =
Идеальные растворы
A …A
B …B
A …B
Eвз(A …A) = Eвз(В …В) = Eвз(A …В)
AиB
Vp-p = VA + VB + …
ΔVсмеш = Vp-p – (VA + VB + …) = 0
73.
ΔUсмеш = Up-p – (UA + UB + …) = 0ΔHсмеш = ΔUсмеш + P× ΔVсмеш = 0
Энтропия и свободная энергия смешения
nB
nA
T = const
изотермический процесс
2
1
Состояние
A
B
Система
×
1
VA, Pt, T
VB, Pt, T
Vt, Pt, T
2
Vt, PA, T
Vt, PB, T
Vt, Pt, T
Vt – конечный объем, Pt – конечное давление
Vt
V
; ΔS B = nB × R× ln t ;
VA
VB
V
V
ΔSсмеш= ΔSA + ΔSB = nA × R× ln t + nB × R× ln t
VA
VB
V
Vt nA + nB
n + nB
1
1
Для идеального газа: t = A
=
;
=
=
nA
ХA
VB
nB
ХB
(закон Авогадро) V A
T= const; ΔSA = nA × R× ln
74.
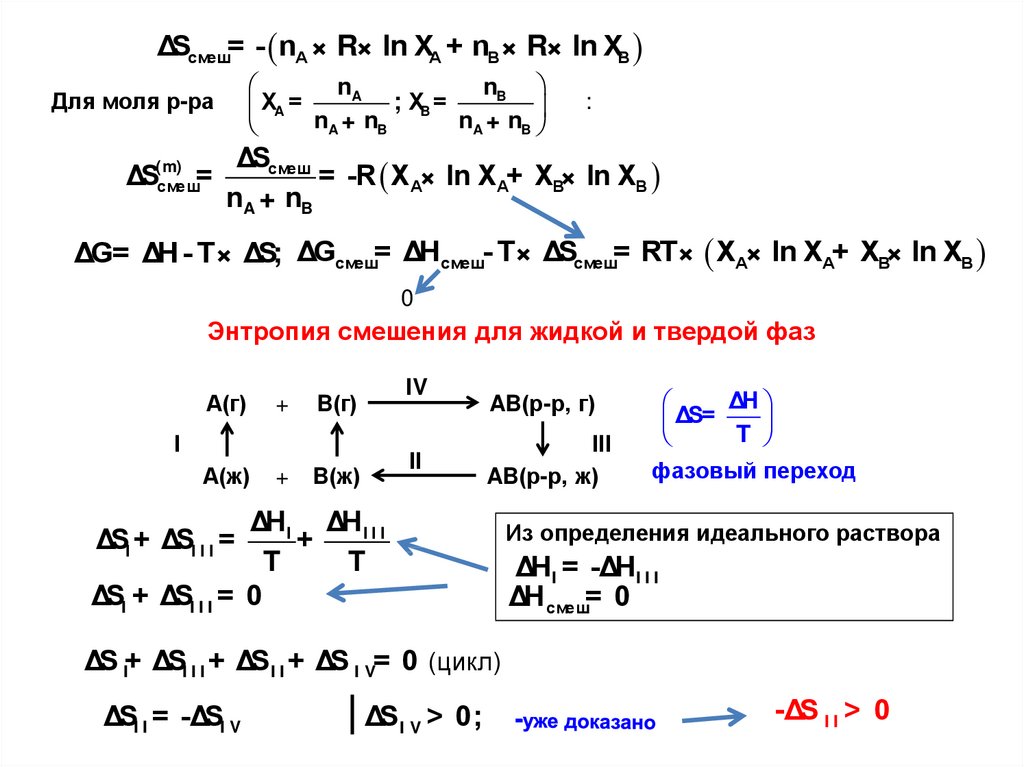
ΔSсмеш= - nA × R× ln ХA + nB× R× ln ХBnA
nB
Х
=
;
Х
=
A
B
nA + nB
nA + nB
Для моля р-ра
ΔS(m)
с ме ш=
:
ΔSсмеш
= -R ХA× ln ХA+ ХB× ln ХB
nA + nB
ΔG= ΔH - T× ΔS; ΔGсмеш= ΔHсмеш- T× ΔSсмеш= RT× ХA× ln ХA+ ХB× ln ХB
0
Энтропия смешения для жидкой и твердой фаз
А(г)
+
B(г)
I
А(ж)
+
B(ж)
IV
AB(р-р, г)
III
AB(р-р, ж)
II
ΔH I ΔH I I I
+
T
T
ΔSI + ΔSI I I = 0
ΔH
Δ
S=
T
фазовый переход
Из определения идеального раствора
ΔSI + ΔSI I I =
ΔHI = -ΔHI I I
ΔH смеш= 0
ΔS I+ ΔSI I I + ΔSI I + ΔS I V= 0 (цикл)
ΔSI I = -ΔSI V
| ΔS
IV
> 0;
-ΔS I I > 0
75.
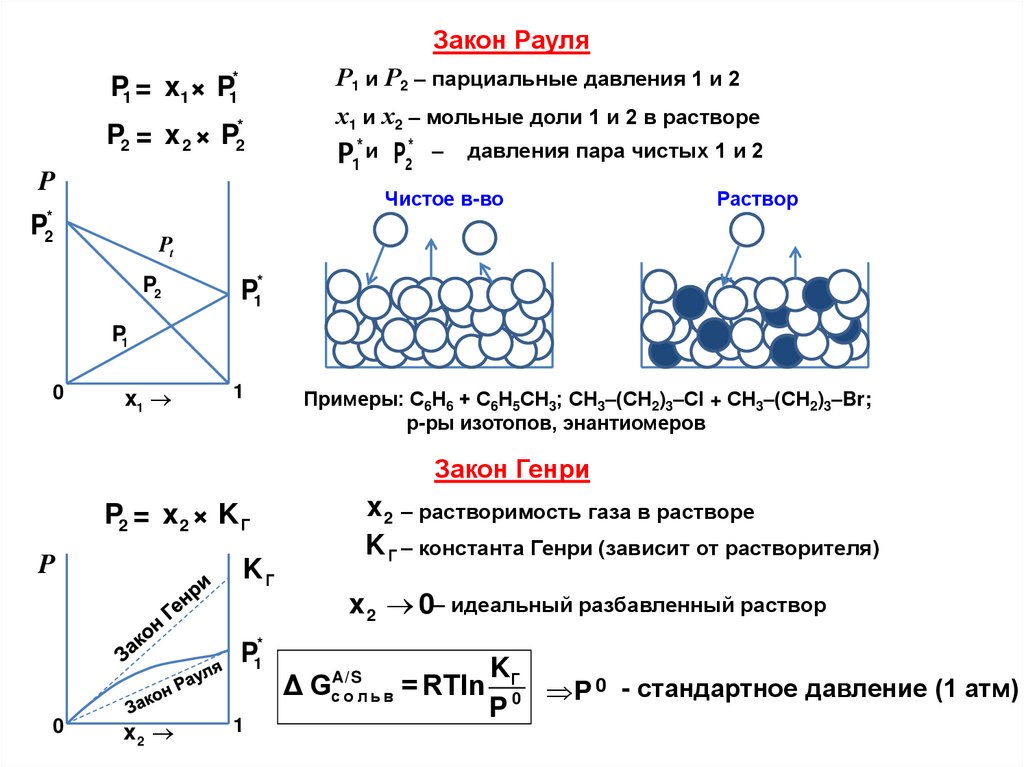
Закон РауляР1 и Р2 – парциальные давления 1 и 2
х1 и х2 – мольные доли 1 и 2 в растворе
P1 = x1 × P1*
P2 = x 2 × P2*
P1* и P2*
P
–
давления пара чистых 1 и 2
Чистое в-во
*
2
P
Раствор
Pt
P2
P1*
P1
0
x1
1
Примеры: С6Н6 + С6Н5СН3; СН3–(СН2)3–Сl + СН3–(СН2)3–Br;
р-ры изотопов, энантиомеров
Закон Генри
x 2 – растворимость газа в растворе
K Γ – константа Генри (зависит от растворителя)
P2 = x 2 × K Γ
P
KΓ
P1*
0
x2
1
x 2 0– идеальный разбавленный раствор
ΔG
A/S
с о льв
KΓ
= RTln 0 P 0 - стандартное давление (1 атм)
P
76.
*P(CH
3 ) 2 CO
Система: Хлороформ + Ацетон
*
PCHCl
3
Cl
Рауль
C
Cl
K Γ( (CH 3 ) 2 CO )
K Γ(CHCl3 )
CH3
H
O
В.С.
Cl
C
CH3
Генри
Парциальные мольные величины
Моля́рный объём — объём одного моля вещества. Величина, получающаяся
от деления молярной массы на плотность. Характеризует плотность упаковки молекул.
V = n1× V1m + n2 × V2m
(идеальный раствор)
V1m и V2m– мольные объемы 1 и 2
Vm (C2 H 5 O H) = 58, 4 с м3
H 2 O+ C2H5 OH
Vm ( H 2 O ) = 18,1 с м3
Σ = 58, 4 см3+ 18,1 см3 = 76, 5 см3 (теория)
74,2 см 3 (эксперимент)
25оС
77.
Для неидеальных растворовV
V = n1× V1m + n2 × V2m + K×
n1× n2
Если K>0 – то Eвз(A …В) < Eвз(A …A) или Eвз(В …В)
силы отталкивания
x1
0
Если K<0 – то Eвз(A …В) < Eвз(A …A) или Eвз(В …В)
силы притяжения
1
Парциальный мольный объем
V = V(n1 , n2 )
dV =
V
×
n
n2 ,P,T
dn1 +
1
dV =
0
V1×
dn2 = V1 × dn1 + V2 × dn2
Vi - const
n1
1
0
2
2
V1 , V2
-парциальные
мольные объемы
ΔV
n2
dn + V × dn
Сравним Vi и
1)
V
×
n
n1 ,P,T
2
Фиксируем состав;
V
(P, T= const)
1 моль A
V = V1× n1 + V2 × n2
0
Vm
Vi = f(n1 , n2 ), Vm = f(n1 , n2 )
р-р AB
78.
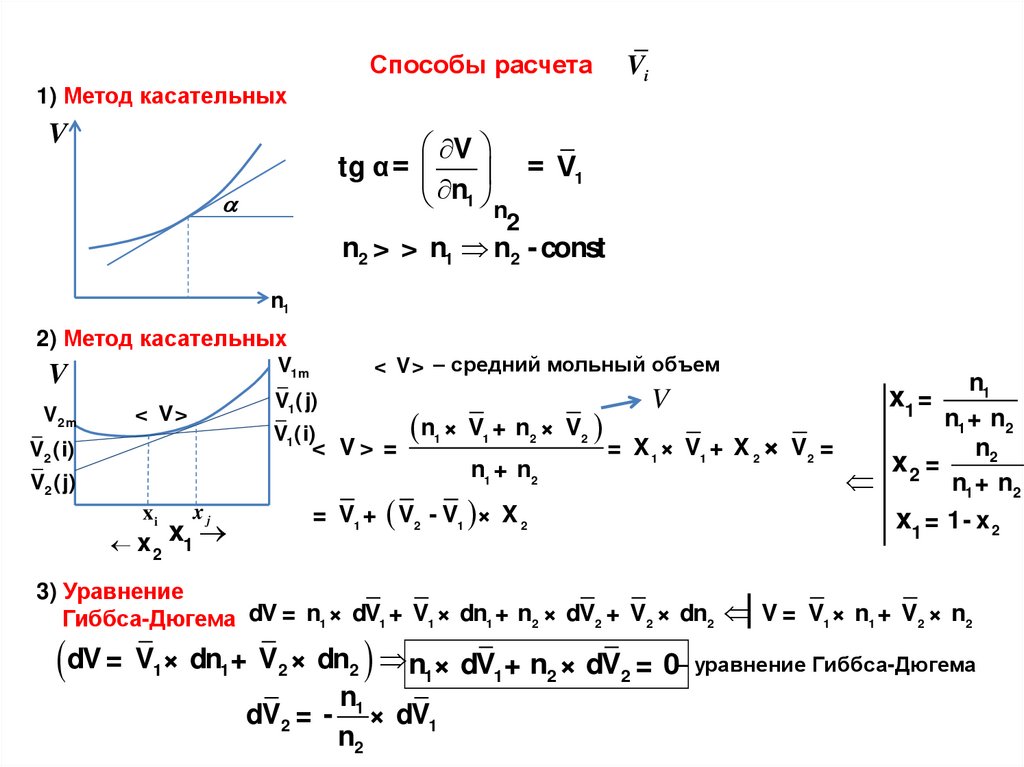
Способы расчета1) Метод касательных
V
Vi
V
tg α =
= V1
n1 n
2
n2 > > n1 n2 - const
n1
2) Метод касательных
V2m
< V> – средний мольный объем
V1m
V
n1
n1 + n2
n1 × V1 + n2 × V2
V1(i)
< V> =
= X 1 × V1 + X 2 × V2 =
n2
x2 =
n1 + n2
n1 + n2
< V>
V2 (i)
V2 (j)
xi
V
V1(j)
= V1 + V2 - V1 × X 2
xj
x 2 x1
x1 =
x1 = 1- x 2
3) Уравнение
Гиббса-Дюгема dV = n1 × dV1 + V1 × dn1 + n2 × dV2 + V2 × dn2
| V = V1 × n1 + V2 × n2
dV = V × dn + V × dn n × dV + n × dV = 0– уравнение Гиббса-Дюгема
1
1
2
2
1
n1
dV2 = - × dV1
n2
1
2
2
79.
Пример: система этанол-водаV
Задача: NaCl в H2O (1000 г)
V(mL) = 1001, 38+ 16, 6253× n2 +
2
+ 1, 7738× n1,5
2 + 0,1194× n2
n2 – молярность NaCl; t=25oC
x эт анол
1)
V(NaCl) = ?
при n2 = 0,50000
2)
V(H 2 O) = ?
при n2 = 0,50000
V
VNaCl =
= 16, 6253+ 1, 7738× 1.5× n2 + 0,1194× 2× n2
n
2 n1
VNaCl (n2 = 0, 50000) = 18,626 mL
Число молей H2O = 1000/18,016 = 55,506
V(mL) = 1001, 38+ 16,6253× 0, 5+ 1,7738× 0, 51,5 + 0,1 194× 0, 5 2 = 1010, 35
V = nH 2 O × VH 2 O + nNaCl × V NaCl = 55, 506× VH 2 O + 9, 313
1010, 35= 55, 506× VH 2 O + 9, 313
VH 2 O = 18, 035 mL
80.
Фундаментальные уравнения для открытых системn × V
H= n × H
V=
i
i
i
i
n × S
A = n × A
S=
i
i
n × U
G= n × G
U=
i
i
dG= -S× dT+ V× dP – закрытые системы
dG= -S× dT+ V× dP+
μ × dn
i
i
– открытые системы
i
G
μ i=
– химический потенциал
n i T,P,n'
i
dU= T× dS- P× dV – закрытые системы
dU= T× dS- P× dV+
U
μi =
n
i S,V,n'
i
μ × dn
i
i
i
– открытые системы
i
i
i
i
81.
dH= T× dS+ V× dP – закрытые системыdH= T× dS+ V× dP+
μ × dn – открытые системы
i
i
i
H
μi =
n
i S,P,n'
i
dA= -S× dT - P× dV – закрытые системы
dA = -S× dT - P× dV+
A
μi =
n
i T,V,n'
μ × dn
i
i
– открытые системы
G f (T , P, ni )
i
i
1
dU= T× dS- P× dV+ μ i × dni |
n
i
i T,P,n'
i
U
S
V
Ui =
=
T×
P×
+ μ i = T× S i- P× Vi + μ i
ni T,P,n'
ni T,P,n'
ni T,P,n'
i
i
i
μi = Ui - T× S+
P× Vi ;
i
μi = Hi - T× Si ;
μi = A i + P× Vi
82.
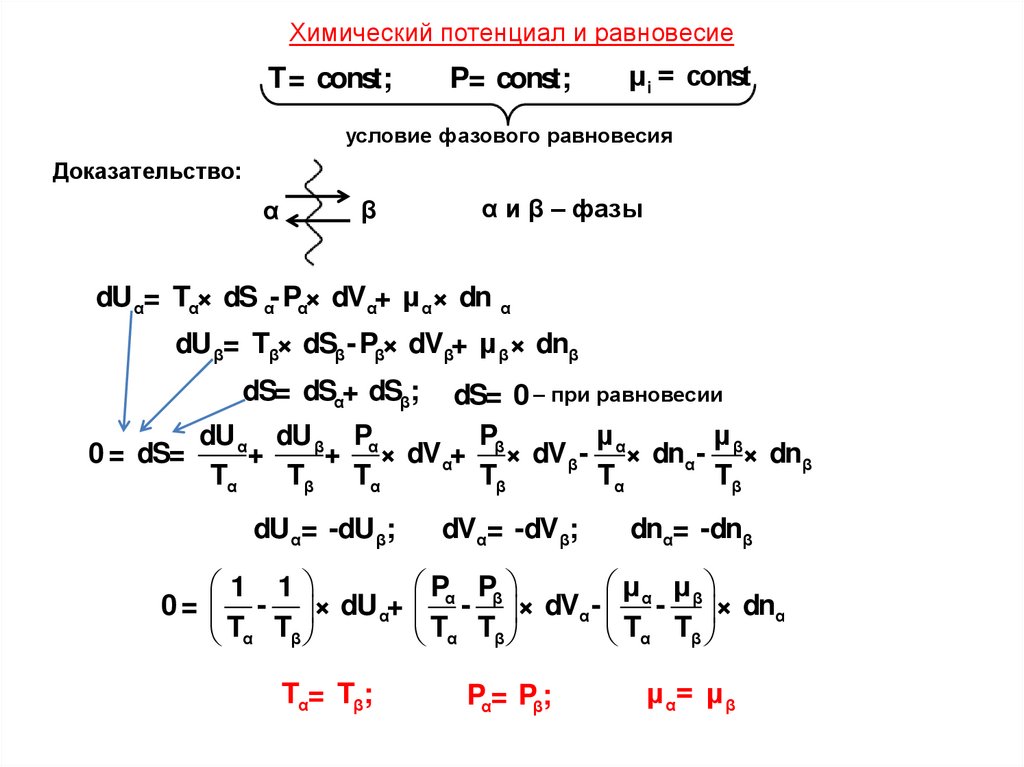
Химический потенциал и равновесиеT= const;
P= const;
μ i = const
условие фазового равновесия
Доказательство:
α
α и β – фазы
β
dU α= Tα× dS α- P×
α dVα+ μ α× dn α
dU β= Tβ× dSβ - P×
β dVβ+ μ β × dnβ
dS= dSα+ dSβ;
dS= 0 – при равновесии
dU α dU β Pα
P
μ
μ
0 = dS=
+
+ × dVα+ β × dVβ - α× dnα - β× dnβ
Tα
Tβ Tα
Tβ
Tα
Tβ
dU α= -dU β;
1 1
0 = - × dU α+
Tα Tβ
Tα= Tβ;
dVα= -dVβ;
dnα= -dnβ
Pα Pβ
μα μβ
×
dV
- × dnα
α
Tα Tβ
Tα Tβ
Pα= Pβ;
μ α= μ β
83.
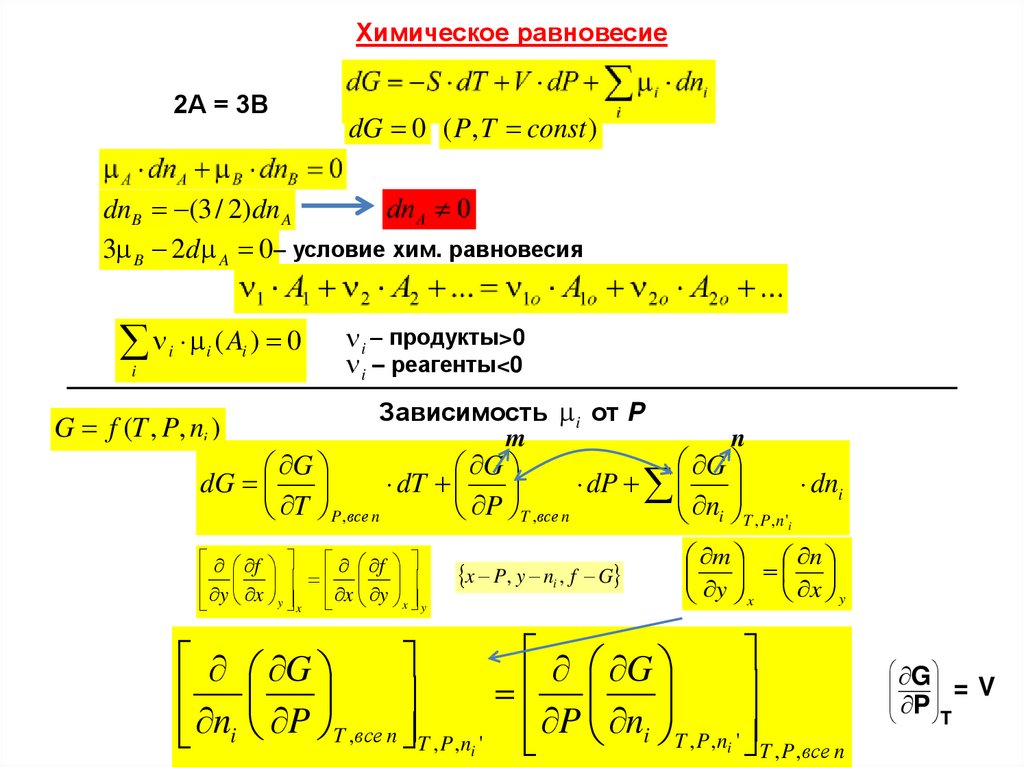
Химическое равновесие2А = 3В
dG 0 ( P, T const )
dnB (3 / 2)dn A
3 B 2d A 0– условие хим. равновесия
(A ) 0
i
i
i
G f (T , P, ni )
i
i – продукты>0
i – реагенты<0
Зависимость i от P
m
n
G
G
G
dG
dT
dP
dni
T P ,все n
P T ,все n
ni T , P ,n 'i
f f
y x y x x y x y
ni
x P, y ni , f G
m
n
y x x y
G
G
P T ,все n T , P ,ni ' P ni T , P ,ni '
T , P ,все n
G =
P
T
V
84.
Vi
Vi
P T ,все n ni T , P ,ni '
P2
2
V dP
i
i
1
P1
Твердые вещества и жидкости
2
V f (P) | слабо |
i ( P2 ) i ( P1 ) Vi ( P2 P1 )
RT
Vi Vi
;
Pi
1
i
Vi
P2
dP
P1
Идеальный
iгаз
P
2
Pi
RT
d i Pi dP 1 d RT P d ln Pi 2 1 RT ln P1
1
ρ P
( P1 1 атм) io
i
1
Неидеальный газ
P(V b) RT
fi – фугитивность (Льюис)
i io RT ln
i io RT ln
P
B ( P 1)
1
fi
1
(исправленное давление) – давление, которое должен иметь реальный газ
при условии его идеального поведения
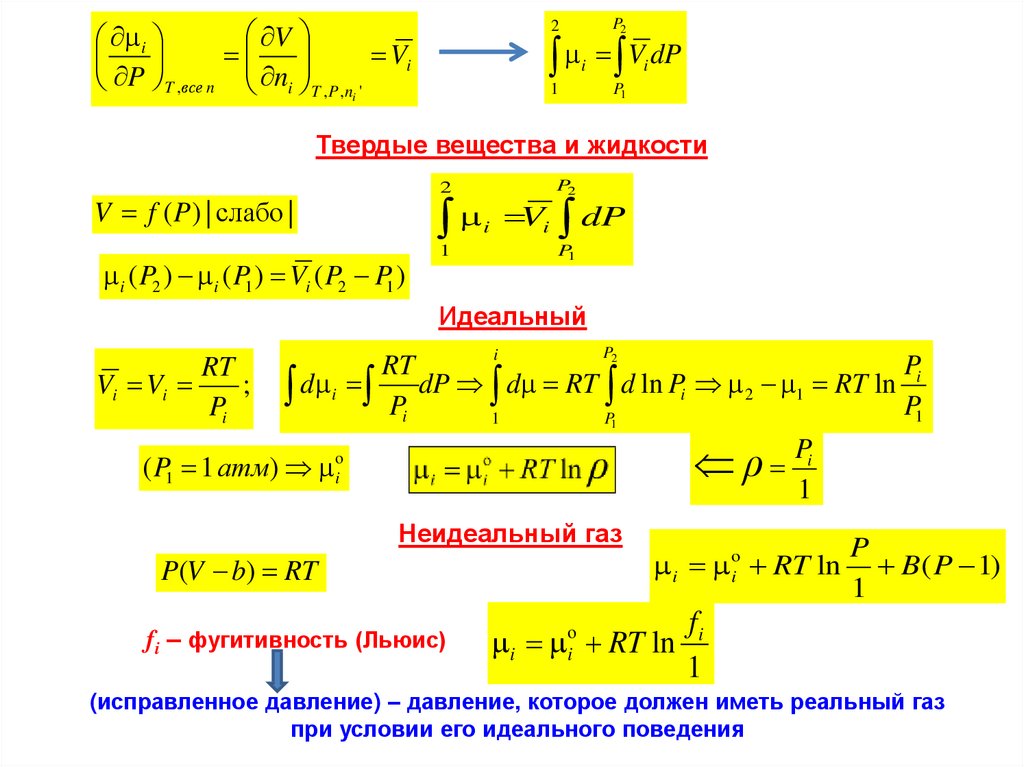
85.
μ = μi
o
i
+ RTln ρ
2
μ i =
1
д.)
μ(и
- μ io = Vi × dP (ид.газ)
i
μi - μio = Vi × dP(реал.газ)
1 VidP
Pi
μ i
= Vi
P
T,всеn
д.)
μ(и
- μi = (Vi - Vi )× dP= α× dP
i
α
P
μ i = μ io + RTlnfi
μ(i ид.) = μ io+ RTlnP
fi
д.)
μ i- μ(и
=
RTln
i
P
f
1
ln i = α× dP
P
RT 0
CO
P = 1200 атм
f = 2663 атм
NH3
P = 1000 атм
f = 204 атм
N2
P = 6000 атм
f = 2·106 атм
86.
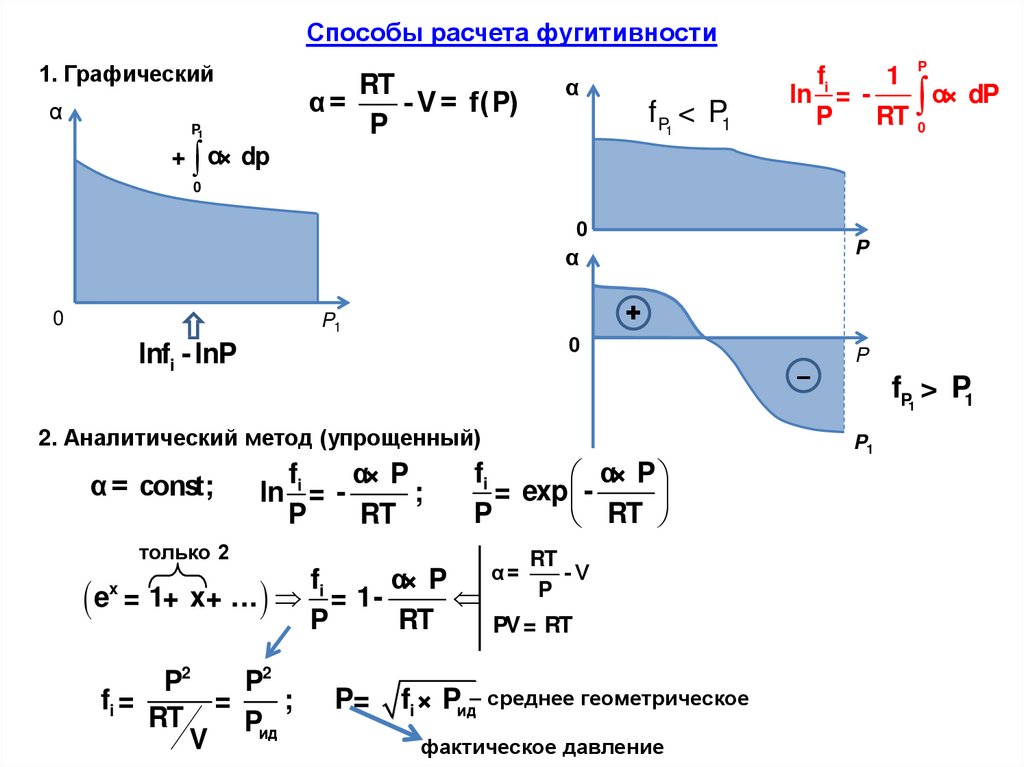
Способы расчета фугитивности1. Графический
α
α=
P1
+ α× dp
RT
- V = f(P)
P
fi
α
fP1 < P1
1
P
ln = α× dP
P
RT 0
0
0
P
α
0
+
P1
0
lnfi - lnP
–
2. Аналитический метод (упрощенный)
α = const;
fi
α× P
ln = ;
P
RT
только 2
fi
α× P
= exp
P
RT
fi
α× P
x
e
=
1+
x+
...
=
1
P
RT
P2
P2
fi =
=
;
RT
Pид
V
P=
α=
RT
-V
P
PV = RT
fi × Pид– среднее геометрическое
фактическое давление
P
fP1 > P1
P1
87.
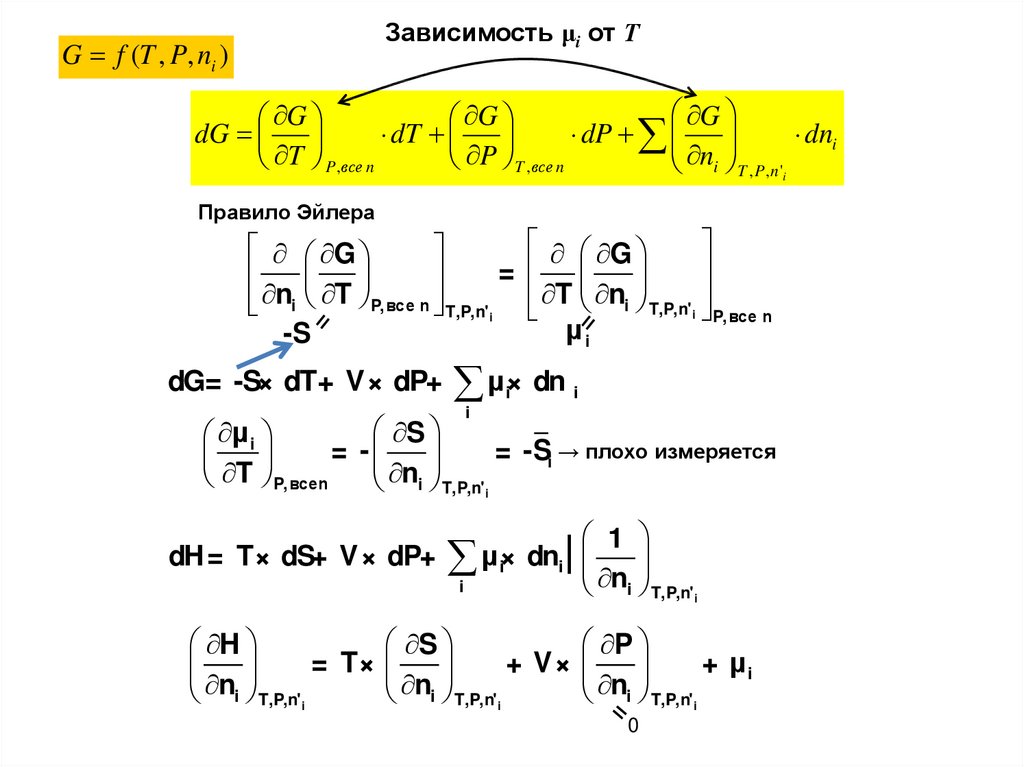
Зависимость μi от ТG f (T , P, ni )
G
G
G
dG
dT
dP
dni
T P ,все n
P T ,все n
ni T , P ,n 'i
Правило Эйлера
G
=
ni T P,все n T,P,n' i
-S
dG= -S× dT+ V× dP+
μ i
=
T P,всеn
G
T ni T,P,n' i P,все n
μi
μ × dn
i
i
i
S
-
= -Si → плохо измеряется
ni T,P,n' i
1
dH= T× dS+ V× dP+ μ i× dni|
n
i
i T,P,n' i
H
S
P
= T×
+ V×
+ μi
n
n
n
i T,P,n'i
i T,P,n' i
i T,P,n' i
0
88.
Hi = T× S+μi
i
μ
i
T = 1 × μ i - μ i | μ i
= -Si
2
T
T T
T
T P,всеn
μ
i
T = 1 × -T× S - μ = - 1 μ + T× S
i
i
i
i
T
T2
T2
μ
i
T = - 1 × H – Уравнение Гиббса-Гельмгольца
i
T
T2
Зависимость μi от ni
P
μ i = μ io + RTln i
G= f(T, P, ni );
1
μi(г аз ) = μi( ж )– при равновесии
Pi = x i × Pi* – закон Рауля
μ i = μ io+ RTlnPi* + RTlnx i
μ*i
μ i = μ + RTlnx i
*
i
G= x1× μ1+ x 2 × μ2 | μ 1(2) = μ
*
– химический потенциал чистой жидкости
+ RTlnx1(2)
1(2)
свободная энергия
смешения
G= x1× μ1*+ x 2 × μ*2+ RT x1× lnx1 + x2 × lnx2
89.
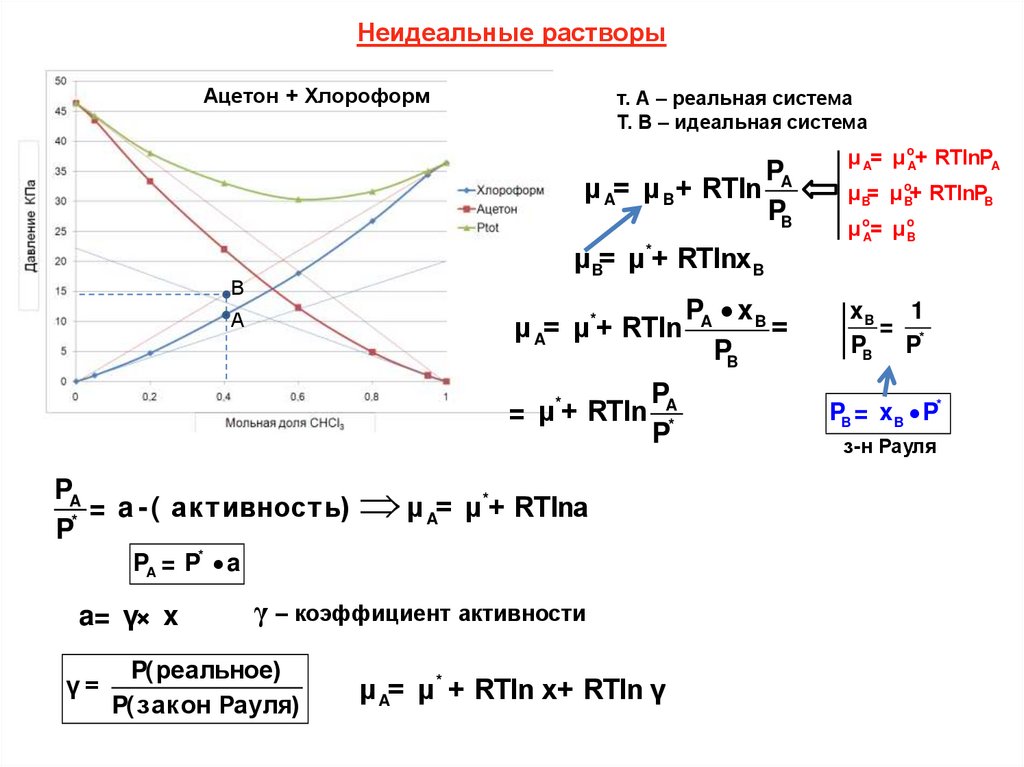
Неидеальные растворыАцетон + Хлороформ
т. А – реальная система
Т. В – идеальная система
PA
μ A= μ B + RTln
PB
μ B= μ*+ RTlnx B
В
А
μ A= μ*+ RTln
PA
= μ + RTln *
P
*
PA
= a - ( а к т ивнос т ь)
*
P
μ =
A
μ*+ RTlna
PA = P* a
a= γ× x
γ=
γ – коэффициент активности
P(ре а льное)
P(з а к он Ра у ля)
μ A= μ * + RTln x+ RTln γ
PA xB
=
PB
μ A= μ oA+ RTlnPA
μ B= μ Bo+ RTlnPB
μ oA= μ Bo
xB 1
= *
PB P
PB = x B P*
з-н Рауля
90.
Теория обратимых процессов в открытых системахα и β – две системы в равновесии
μα μ β
=
;
T
T
d
d
μα
μ
= d β
T
T
обратимые процессы
два равновесных состояния
μi/ T
μ i/ T
μi μi/ T
=
×
dP+
×
dT+
× dni
T P T,всеn
T P,всеn
ni T,P,n' i
μi/ T
Vi
μ i
×
dP=
×
dP
=
i
T
P T,всеn
P T,всеn
μi/ T
× dni = Rdlnai
ni T,P,n' i
μi/ T
Hi
×
dT
=
× dT
2
T
T P,всеn
d
V
= Vi
ni T,P,n' i
μ i= μ*i+ RTlnx i
Pi = a× Pi*
dμ*i= 0; μ i= μ*i+ RTlnai
уравнение Гиббса-Гельмгольца
μ i Vi
H
=
× dP- 2i × dT+ Rdlnai – общее уравнение химического потенциала
T
T
T
91.
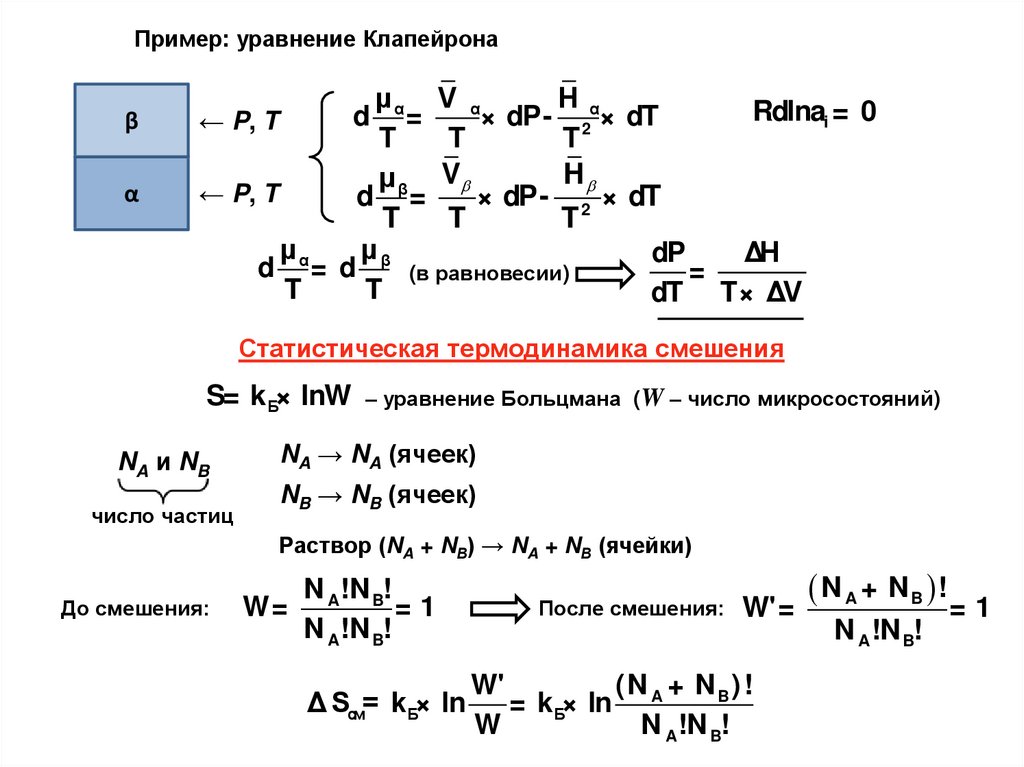
Пример: уравнение Клапейронаβ
α
μα V α
Hα
Rdlnai = 0
=
×
dP× dT
← P, T
2
T
T
T
H
μ β V
← P, T
d =
× dP- 2 × dT
T
T
T
μ
μ
dP
ΔH
d α = d β (в равновесии)
=
T
T
dT T× ΔV
d
Статистическая термодинамика смешения
S= kБ× lnW
NA и NB
число частиц
– уравнение Больцмана (W – число микросостояний)
NA → NA (ячеек)
NB → NB (ячеек)
Раствор (NA + NВ) → NA + NВ (ячейки)
До смешения:
N !N !
W= A B = 1
N A !N B!
После смешения:
W'=
( N A + N B) !
W'
Δ Sсм= k Б× ln
= k Б× ln
W
N A !N B!
NA+
N B !
=1
N A!N B!
92.
lnN! » NlnN - N – приближение Стирлинга( N + N B) !
ΔSсм= k Б× ln A
= k Б× ln( N A + N B ) ! - lnN A! - lnN B! =
N A !N B!
= k Б× ( N A + N B ) ln( N A + N B ) - ( N A + N B ) - N A lnN A - N A - N BlnN B - N B =
= k Б× N A ln( N A + N B )+ N Bln( N A + N B ) - N A lnN A - N BlnN B =
N + NB
N + NB
= k Б× N A ln A
+ N Bln A
= -k Б×
NA
NB
xA
NA
NB
N
ln
+
N
ln
B
A
N
+
N
N
+
N
A
B
A
B
xA
xB
xB
NA
N + NB
NB
N + NB
ΔSсм (на 1 моле к у лу) = -k Б×
ln A
+
ln A
=
NA
N A + NB
NB
N A + NB
= -k Б× x A lnx A + x BlnxB
k Б=
R
; N Ав – число Авогадро
N Ав
ΔSсм (на 1 моль раст вора) = -R× x A lnx A + xBlnxB
93.
Коллигативные свойства растворовЗависят от числа частиц, но не от их природы.
Раствор: 1) летучий растворитель и нелетучее
вещество
при предельном разбавлении (Pобщ = Рр-ль);
2) растворенное вещество не растворяется в
твердом растворителе.
Рo – атмосферное давление
P
1 атм
Tк – нормальная т. кипения
тв.
Tпл - δT
Р-р
Tпл
ж.
P’ – пониженное давление
Po , Tк
Po , Tк+ ΔTк
Р-ль
ΔTк – повышение температуры кипения
Tпл – нормальная температура
плавления
Pc , Tc
P", Tc
δT – понижение температуры
P ', Tb
Р-р
плавления
газ
P' c , Tc - ΔTf
T
Pc, Tc – давление и температура в
тройной точке
P’c – понижение давления в тройной
точке
ΔTf – понижение температуры в
тройной точке
P” – пониженное давление при
температуре тройной точки
94.
1. Повышение точки кипенияPo ΔH исп(1)
P'
×
x1 = o - з а к он Ра у ля ln =
P'
R
P
1
1
T
T
+
Δ
T
к к
к
W × M1
-lnx1 = -ln(1- x 2 ) x 2 = 2
M 2 × W1
-lnx1 =
1
Ур-ние Клаузиуса-Клапейрона
1
(пунктирная линия)
T
T
+
Δ
T
к к
к
ΔH исп(1)
×
R
ln( 1- x)
-x ( x< < 1 )
n2
n1 + n2
n2
( n2 < < n1 )
n1
W2 и W1 – массы растворенного вещества и растворителя
M2 и M1 – молекулярные массы растворенного вещества и растворителя
ΔH исп(1)
×
R
1
ΔH исп(1) ΔTк
W2 × M 1 × R× Tк2
1
× 2 ; ΔTк =
=
M 2 × W1 × ΔH исп(1)
R
Tк
Tк Tк+ ΔTк
ΔTк = K в× m2
m2 =
W2
– моляльность растворенного вещества
M 2 × W1
(W1 >> W2)
K в – эбулиоскопическая константа
R× Tк2 × M 1
K в=
ΔH исп(1)
95.
2. Понижение точки замерзанияln
P" ΔH исп( 1)
=
×
P'
R
=
ΔH субл( 1)
×
R
ln
1
1
=
Tпл - ΔTf Tпл
1
1 ΔH пл( 1)
×
R
Tпл - ΔTf Tпл
PC
ΔH субл(1)
=
×
P' C
R
1
1
T
Δ
T
T
пл
f
пл
P"× P' C
ΔH пл(1)
ln
= lnx1 = ×
P' C × PC
R
Аналогично:
Ур-ние Клаузиуса-Клапейрона
(пунктирная линия)
1
1
ΔH исп= ΔH субл - ΔH пл
T
Δ
T
T
пл
f
пл
Ур-ние Клаузиуса-Клапейрона
для сублимации
1
1
T
Δ
T
T
пл
f
пл
P" = x - з а к он Ра у ля
PC 1
ΔTf = K f × m2 m2 – моляльность растворенного вещества
R× Tп2л× M 1
– криоскопическая константа
Kf =
ΔH исп(1)
96.
3. Идеальная растворимостьА(ж)
А(тв)
→μi (ж)
→μi (тв)
При равновесии:
μ*A ( т в) = μ A ( ж ) = μ*A ( ж )+ RTlnx A
x A = ? при Т
*
lnx A = - μ*A ( ж ) - μ(
т в) / RT =
lnx A =
μ* A (т в) = μA (ж )
-ΔGпл( T) ΔGпл( Tпл)
+
RT
RTпл
|
ΔGпл(Tпл) = 0
-ΔGпл( T)
RT
ΔGпл( Tпл)
= 0
Tпл
ΔH пл 1 1
lnx A =
-
R Tпл T
x A = exp
Δ H пл (T - Tпл)
RTплT
T Tпл
x 1
97.
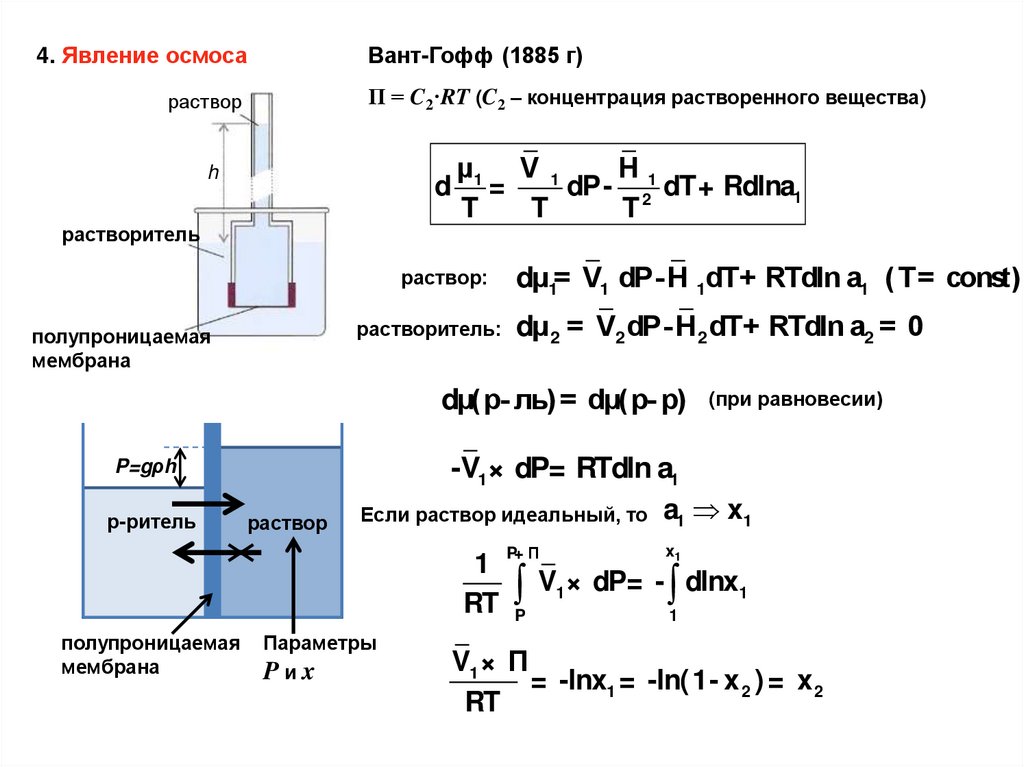
4. Явление осмосаВант-Гофф (1885 г)
П = C2·RT (C2 – концентрация растворенного вещества)
раствор
h
d
μ1 V 1
H
=
dP- 21 dT+ Rdlna1
T
T
T
растворитель
раствор:
растворитель:
полупроницаемая
мембрана
dμ1= V1 dP-H 1dT+ RTdln a1 (T= const)
dμ 2 = V2 dP-H 2 dT+ RTdln a2 = 0
dμ(р- ль) = dμ(р- р)
P=gρh
р-ритель
раствор
-V1× dP= RTdln a1
Если раствор идеальный, то a1 x1
1
RT
полупроницаемая
мембрана
(при равновесии)
Параметры
Pиx
P+ Π
x1
V × dP= - dlnx
1
P
1
1
V1 × Π
= -lnx1 = -ln( 1- x 2 ) = x 2
RT
98.
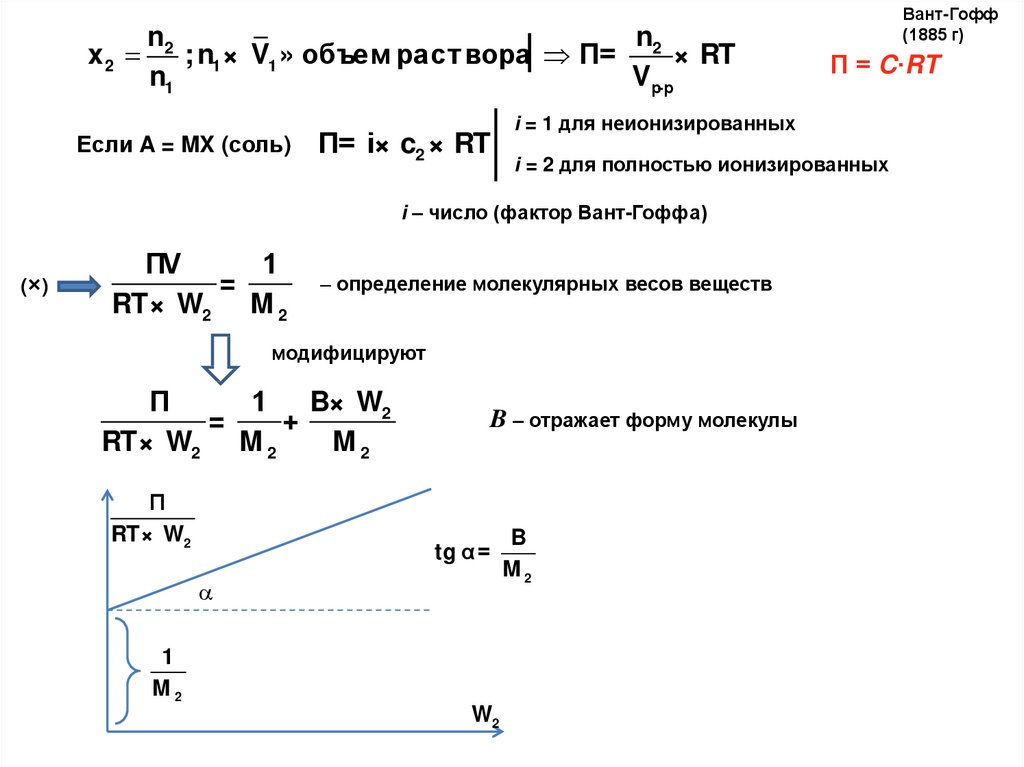
x2n
n2
; n1 × V1 » объе м ра с т вора| Π= 2 × RT
Vр-р
n1
Если A = MX (соль)
Π= i× c2 × RT
ΠV
1
=
RT× W2 M 2
i = 2 для полностью ионизированных
– определение молекулярных весов веществ
модифицируют
B× W2
Π
1
=
+
RT× W2 M 2
M2
Π
RT× W2
B – отражает форму молекулы
tg α =
1
M2
W2
П = C·RT
i = 1 для неионизированных
i – число (фактор Вант-Гоффа)
(× )
Вант-Гофф
(1885 г)
B
M2
99.
Правило фазТД система – гетерогенная или гомогенная
Гетерогенная – есть поверхность раздела
Гомогенная – нет поверхности раздела
Фаза – часть гетерогенной системы, ограниченной поверхностью раздела
и обладающей одинаковыми физическими свойствами
Степень свободы – число независимых интенсивных параметров, необходимых
для описания системы
f = f(P, Vm , T);
Vm = f(P, T) – 2 степени свободы
F = (полное число независимых параметров) –
– (число ограничений, накладываемых на систему)
С – число компонентов
Ф – число фаз
Интенсивные параметры: P, T и Ф·(С–1) мольных долей
Число ограничений:
μiα = μiβ
С·(Ф–1)
Ф – число фаз
F = 2 + Ф·(С–1) – С(Ф-1) = 2 + С – Ф
F=2+С–Ф
– правило фаз Гиббса
100.
PyБ x Б× PБ*
=
yT x T × PT*
ж
газ + ж
x'
Pa
a
y'
F = 2 + С – Ф; F = 4 – Ф
F = 3 – Ф (T или P = const)
газ
Пусть P = 1 атм |
xT=1
yT=1
xБ = f(T)
x Б× PБ*
yБ=
1
1- PТ*
x Б= * *
PБ - PT
zБ(а)
xБ=1
yБ=1
P= PT* + x Б(PБ* - PT* ) PТ* и PБ* f( T)
– Клапейрон
Число молей в каждой фазе?
xБ zБ yБ – стягивающая линия
газ + ж
102oC
zБ
T1
газ
T
nБ – число молей бензола;
n – полное число молей бензола и толуола
yБ
xБ
80oC
zБ – мольная доля бензола в системе
nБ= zБ× n( г аз )+ n(ж)
ж
xT=1
yT=1
nБ= yБ× n( г а з )+ x Б× n( ж )
zБ
Правило рычага:
xБ=1
yБ=1
n(г аз )× (zБ- yЬ ) = n(ж)× (x Б- zЬ )
n( г а з ) x Б- zБ
=
n( ж)
zБ - y Б
101.
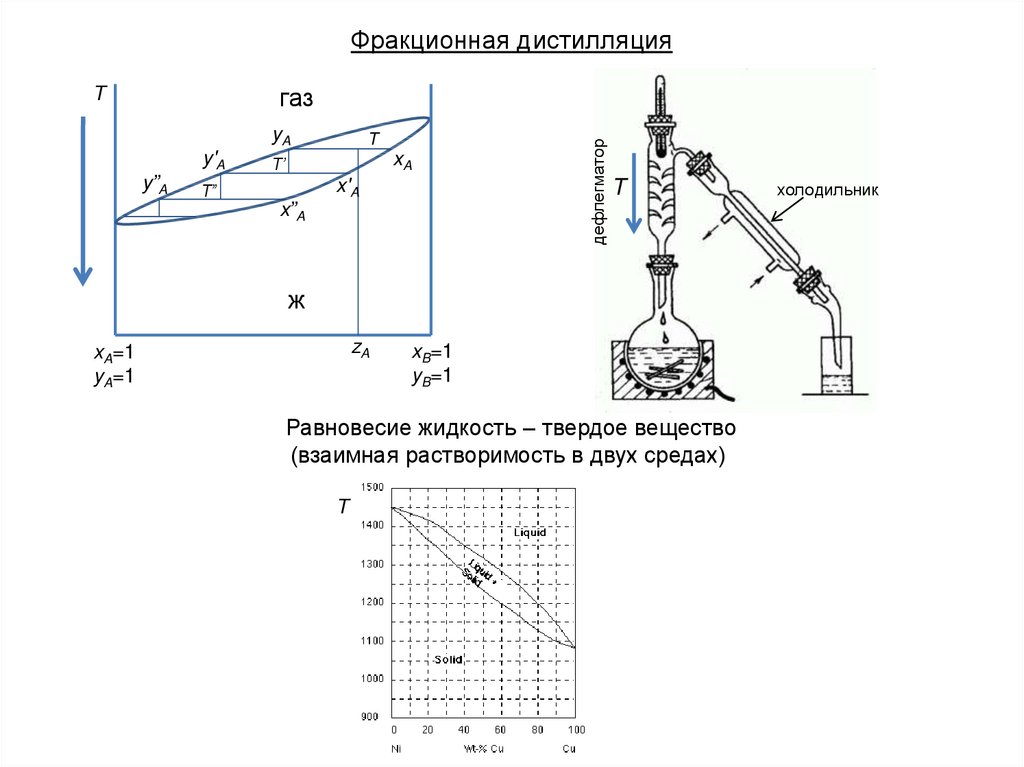
Фракционная дистилляцияyA
y'A
y”A
T
xA
T’
x'A
T”
x”A
дефлегматор
газ
T
T
ж
zA
xA=1
yA=1
xB=1
yB=1
Равновесие жидкость – твердое вещество
(взаимная растворимость в двух средах)
T
холодильник
102.
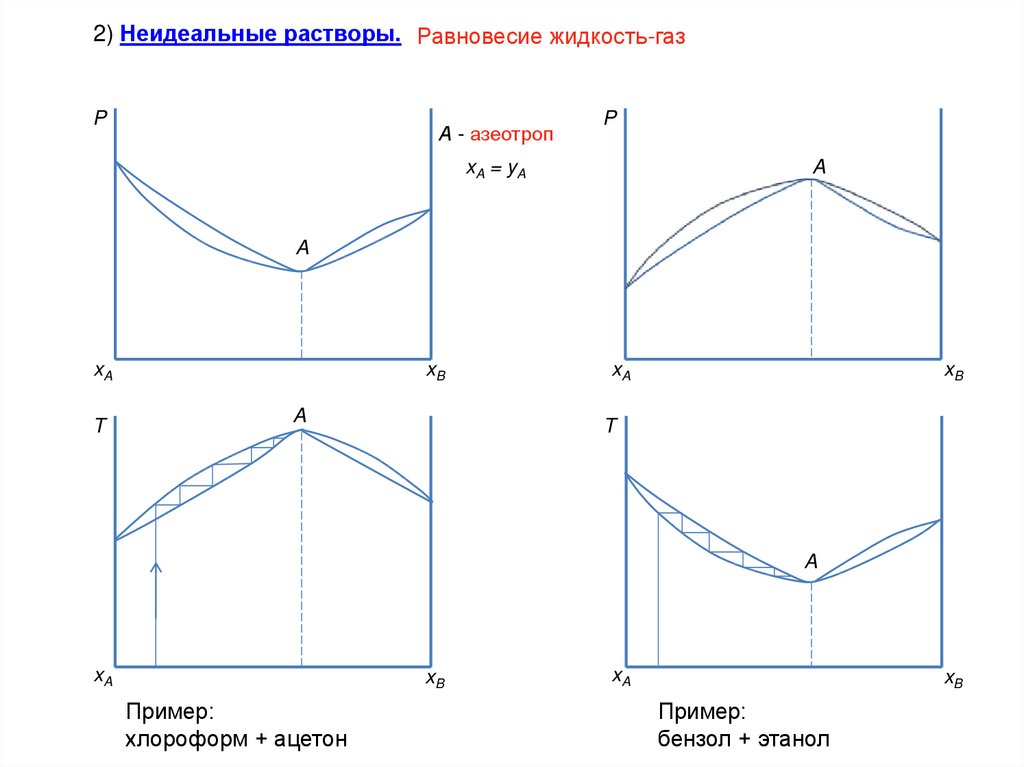
2) Неидеальные растворы. Равновесие жидкость-газP
А - азеотроп
P
xA = yA
A
A
xA
T
xB
A
xA
xB
T
A
xA
xB
Пример:
хлороформ + ацетон
xA
xB
Пример:
бензол + этанол
103.
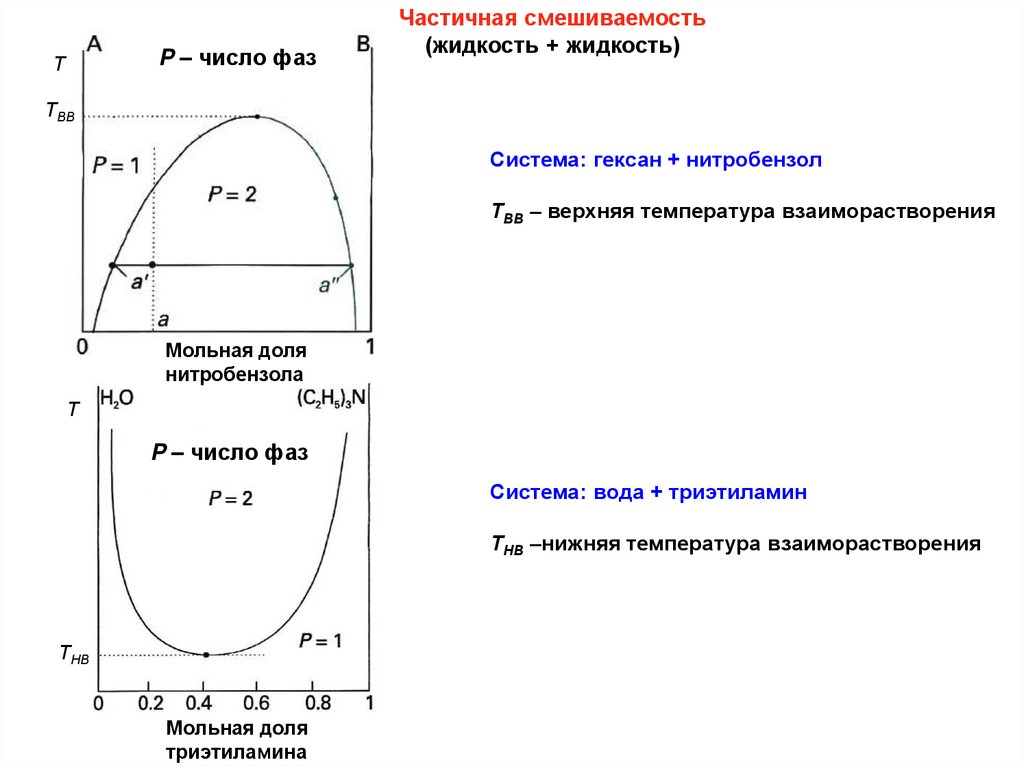
P – число фазТ
Частичная смешиваемость
(жидкость + жидкость)
ТВВ
Система: гексан + нитробензол
ТВВ – верхняя температура взаиморастворения
Мольная доля
нитробензола
Т
P – число фаз
Система: вода + триэтиламин
ТНВ –нижняя температура взаиморастворения
ТНВ
Мольная доля
триэтиламина
104.
Система: вода + никотинТВВ
Т
ТНВ
Мольная доля
никотина
105. Перегонка частично смешивающихся жидкостей
Тпар
жидкость
жидкость
состав
106.
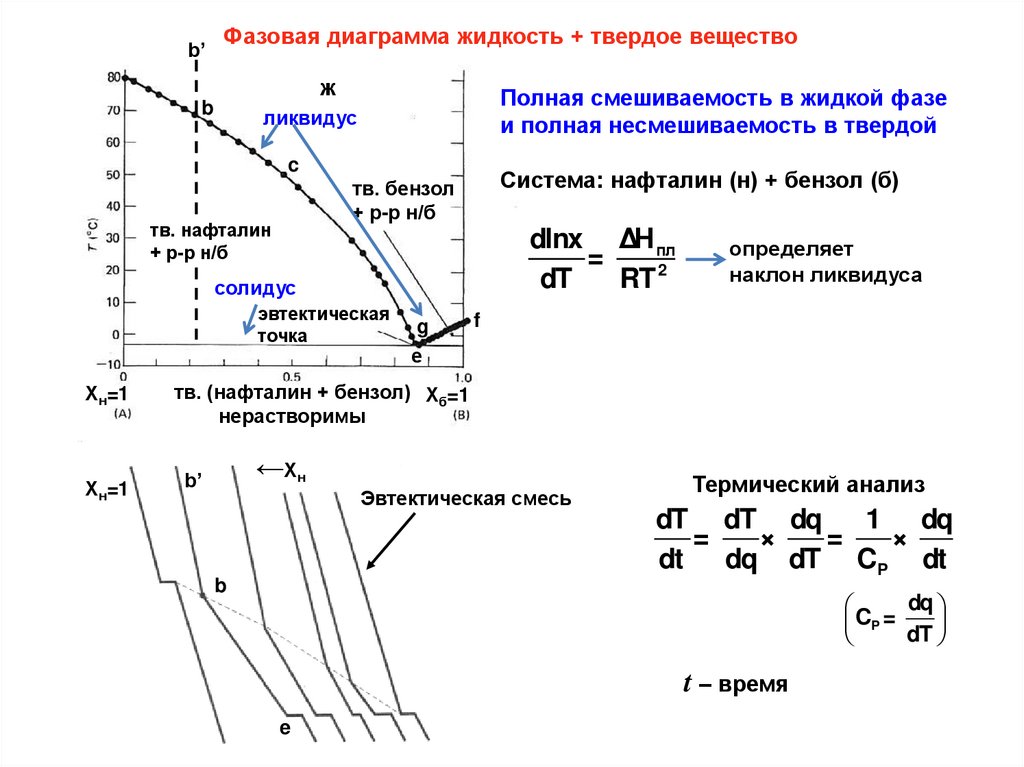
b’Фазовая диаграмма жидкость + твердое вещество
ж
b
Полная смешиваемость в жидкой фазе
и полная несмешиваемость в твердой
ликвидус
c
Система: нафталин (н) + бензол (б)
тв. бензол
+ р-р н/б
тв. нафталин
+ р-р н/б
dlnx ΔH пл
=
dT
RT 2
солидус
эвтектическая
точка
Хн=1
Хн=1
g
е
определяет
наклон ликвидуса
f
тв. (нафталин + бензол) Хб=1
нерастворимы
←Хн
b’
Эвтектическая смесь
b
Термический анализ
dT dT dq
1 dq
=
×
=
×
dt dq dT CP dt
dq
CP =
dT
t – время
e
107.
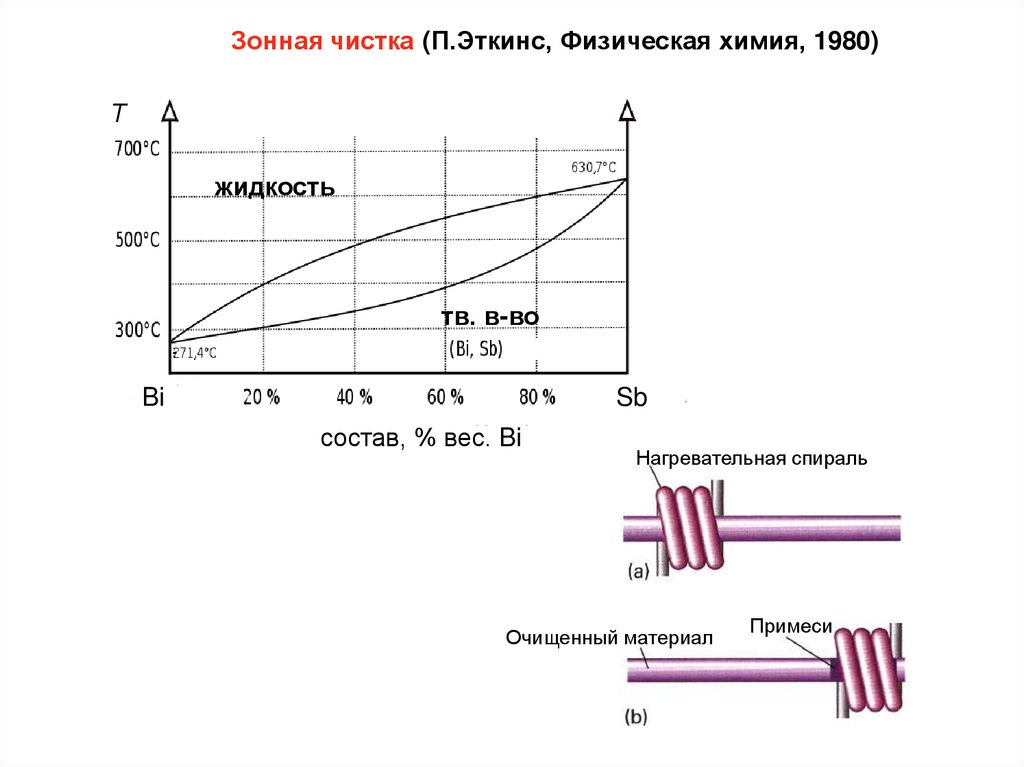
Зонная чистка (П.Эткинс, Физическая химия, 1980)Т
жидкость
тв. в-во
Bi
Sb
состав, % вес. Bi
Нагревательная спираль
Очищенный материал
Примеси
108.
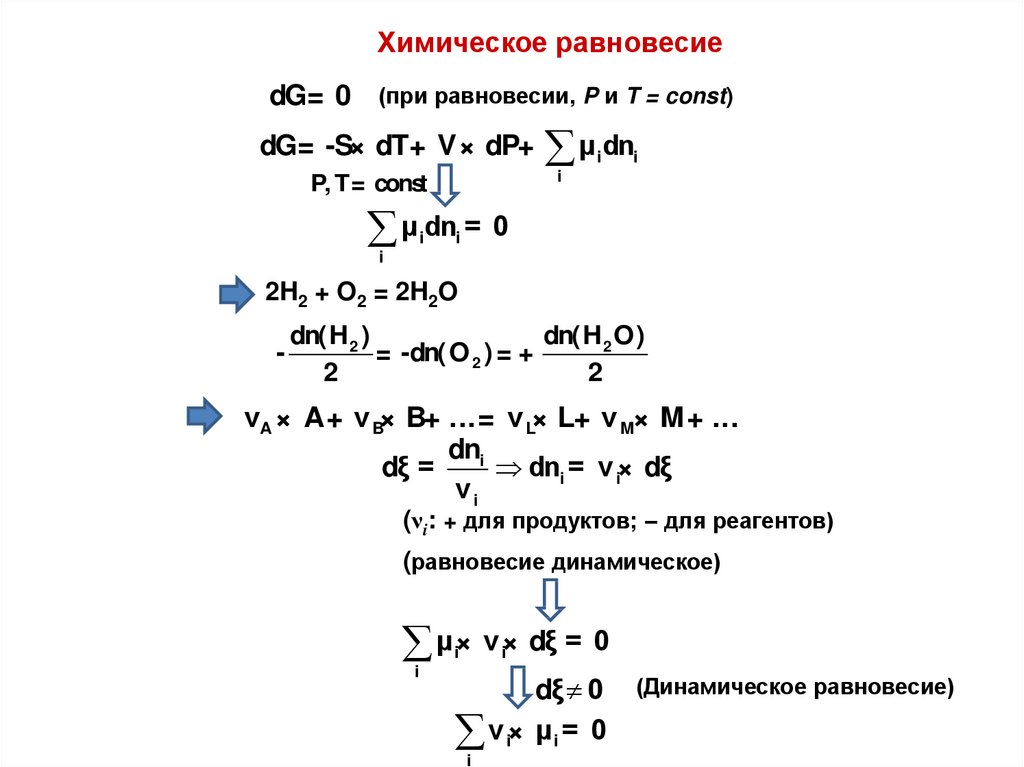
Химическое равновесиеdG= 0 (при равновесии, P и T = const)
dG= -S× dT+ V× dP+
μ dn
i
i
i
P, T= const
μ dn = 0
i
i
i
2H2 + O2 = 2H2O
-
dn(H 2 )
dn(H 2 O)
= -dn(O 2 ) = +
2
2
νA × A+ ν B× B+ ...= ν L× L+ ν M× M+ ...
dni
dξ =
dni = ν i× dξ
νi
(νi: + для продуктов; – для реагентов)
(равновесие динамическое)
μ × ν × dξ = 0
i
i
i
dξ 0
ν i× μi = 0
i
(Динамическое равновесие)
109.
Pi - па рциа льное да вле ние iPi
μ i= μ + RTln o
Pi
o
i
μ io- с т а нд. ХП при Pio = 1 а т м
Pi
ρi= o
Pi
μi= μ + RTlnρi ;
o
i
o
ν
×
μ
i i = -RT ν i× lnρi | ν i× μi= 0
i
i
i
-RT ν i× lnρi = -RTln ρi
i
ρ
νi
i
=
i
ρ
ρ
νL
ρ
×ρ
×
L
νA
A
ν× μ
i
o
i
ν Mi
M
B
νB
νi
Π- произ ве де ние
alnx= lnx a
...
...
= ΔG po– стандартная свободная энергия реакции
i
ΔG po= ν L× μ Lo+ νM × μ oM+ ... - νA × μ oA+ ν B× μBo+ ...
ν× μ
i
ρ
i
o
i
– не зависит от давления !!!
i
νi
= Kp
– (const при T = const)
i
K p– константа равновесия
ΔG po = -RTlnK
p
110.
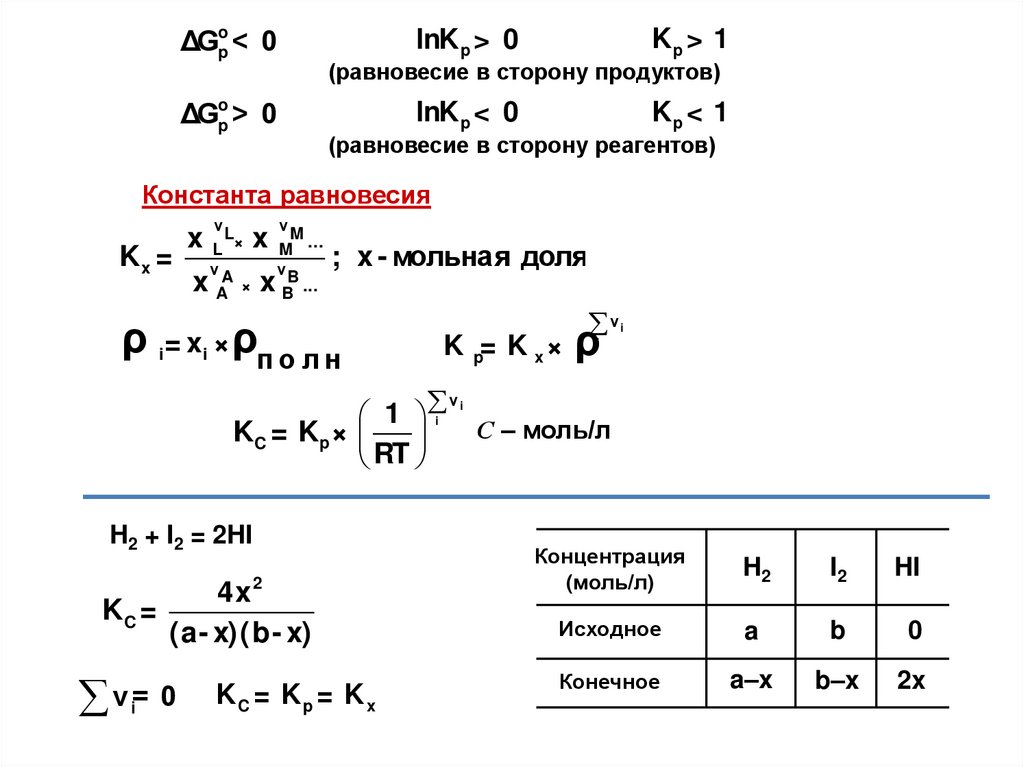
ΔGpo < 0ΔGpo > 0
Kp > 1
lnK p > 0
(равновесие в сторону продуктов)
Kp < 1
lnK p < 0
(равновесие в сторону реагентов)
Константа равновесия
Kx =
ρ
i
x
x
νL
L ×
νA
A ×
x
x
νM
M ...
νB
B ...
; x - мольна я доля
= xi × ρ
по лн
K p= K x ×
νi
ρ
i
i
1 i С – моль/л
KC = Kp ×
RT
ν
H2 + I2 = 2HI
2
KC =
4x
(a- x)(b- x)
ν i= 0
KC = Kp = K x
Концентрация
(моль/л)
H2
I2
HI
Исходное
a
b
0
Конечное
a–x
b–x
2x
111.
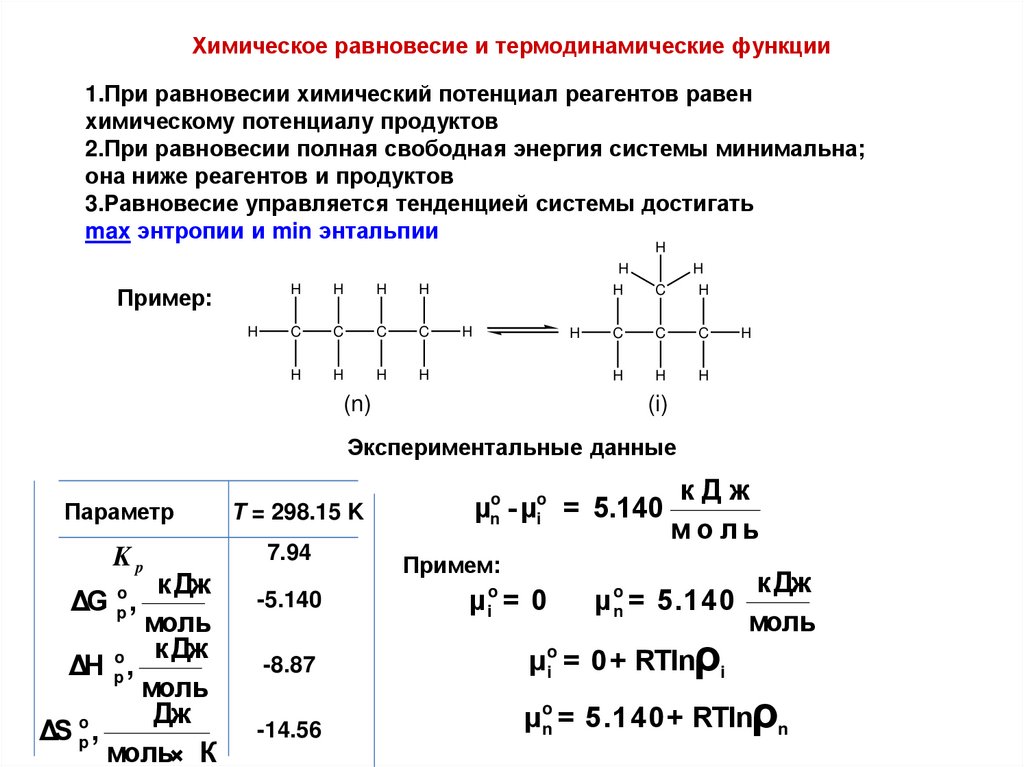
Химическое равновесие и термодинамические функции1.При равновесии химический потенциал реагентов равен
химическому потенциалу продуктов
2.При равновесии полная свободная энергия системы минимальна;
она ниже реагентов и продуктов
3.Равновесие управляется тенденцией системы достигать
max энтропии и min энтальпии
H
Пример:
H
H
H
H
H
C
C
C
C
H
H
H
H
H
H
(n)
H
H
C
H
H
C
C
C
H
H
H
H
(i)
Экспериментальные данные
Параметр
Kp
к Дж
моль
к Дж
ΔH po ,
моль
Дж
ΔS po ,
моль× К
ΔG po ,
T = 298.15 K
7.94
-5.140
μno - μio = 5.140
кДж
мо ль
Примем:
μ io = 0
μ no = 5.140
к Дж
моль
-8.87
μio = 0+ RTlnρi
-14.56
μno = 5.140+ RTlnρn
112.
1. При равновесии химический потенциал реагентов равенхимическому потенциалу продуктов
μ RT
ρ = ρ= 1 ат м (начальное состояние)
ρ = 1.78 ат м; ρ = 0.22 ат м (конечное состояние)
n-C4H10
n
n
i
нач.
i-C4H10
кон.
K p=
кон.
нач.
ln ρ
i
2-x
( x - па рциа льное да вле ние n-C4H10)
x
μ n= 1 .4 к Дж
μ i = 1.4 к Дж
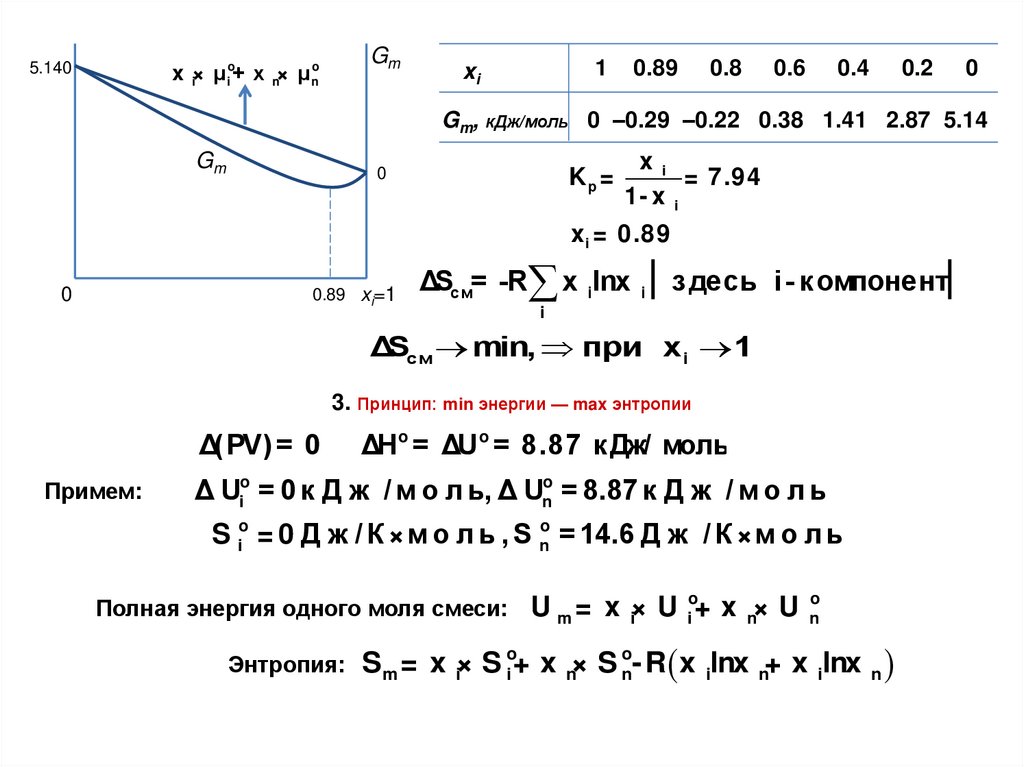
2. Минимум свободной энергии
G m= xi ×μi + xn ×μn Gm - молярная э нергия Гиббса
P
P
x i= i ; x n= n ; P- общ е е да вле ние
P
P
μ i= μ io + RTlnPi = μ io + RTlnx i+ RTlnP
μ n= μno +RTlnxn +RTln P
G m= xi × μio + xn× μno + RTlnP+ RT xilnx i+ xnlnx
n
0
Примем:
μ io = 0
μ no = 5.140 к Дж / моль
И P=1
ат м
113.
5.140Gm
x i× μ io+ x n× μ no
1
xi
0.89
0.8
0.6
0.4
0.2
0
Gm, кДж/моль 0 –0.29 –0.22 0.38 1.41 2.87 5.14
Gm
0
Kp =
xi
= 7.94
1- x i
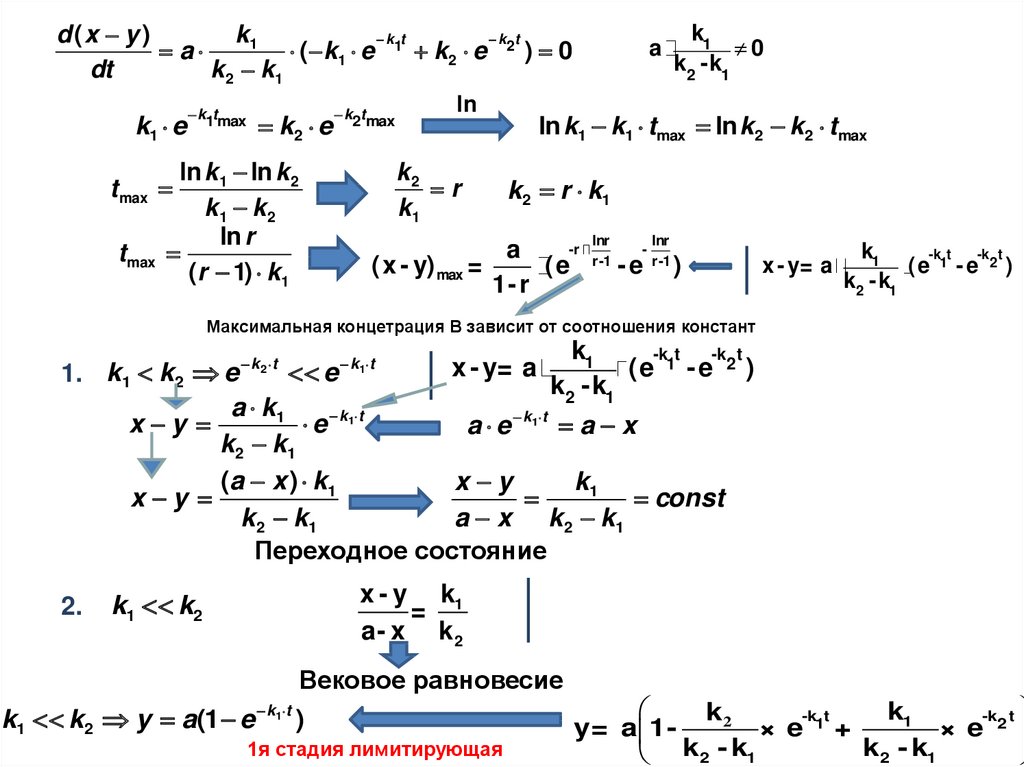
x i = 0.89
0
0.89 xi=1
ΔSсм= -R x ilnx
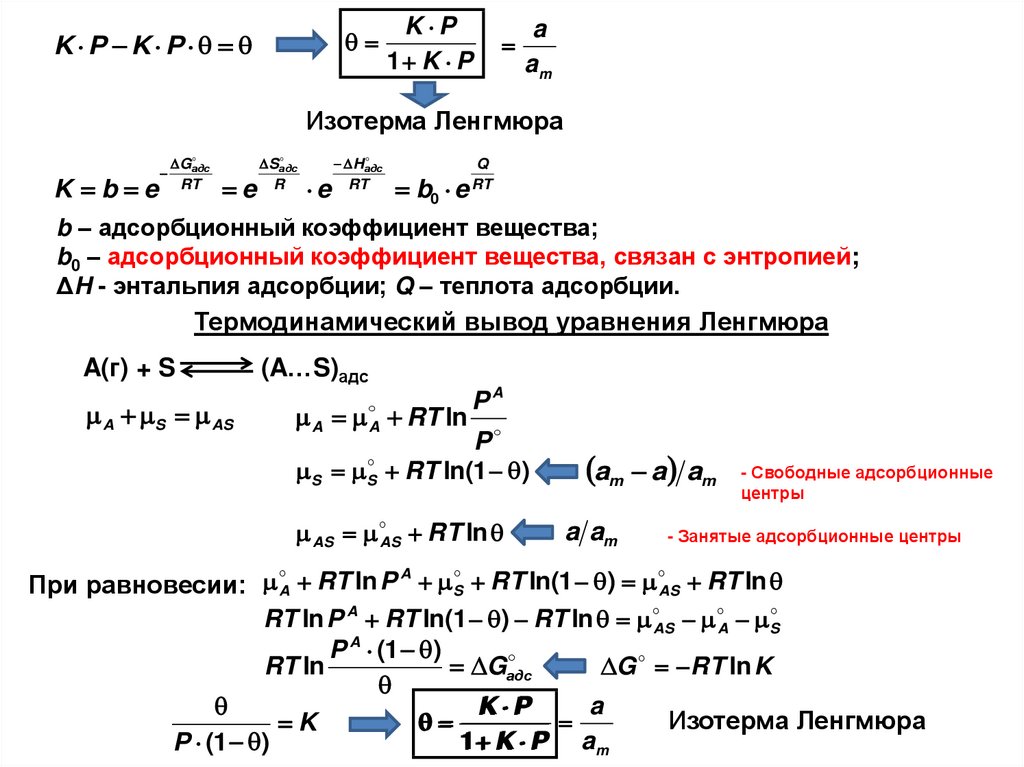
i
| з десь
i - к омпоне нт|
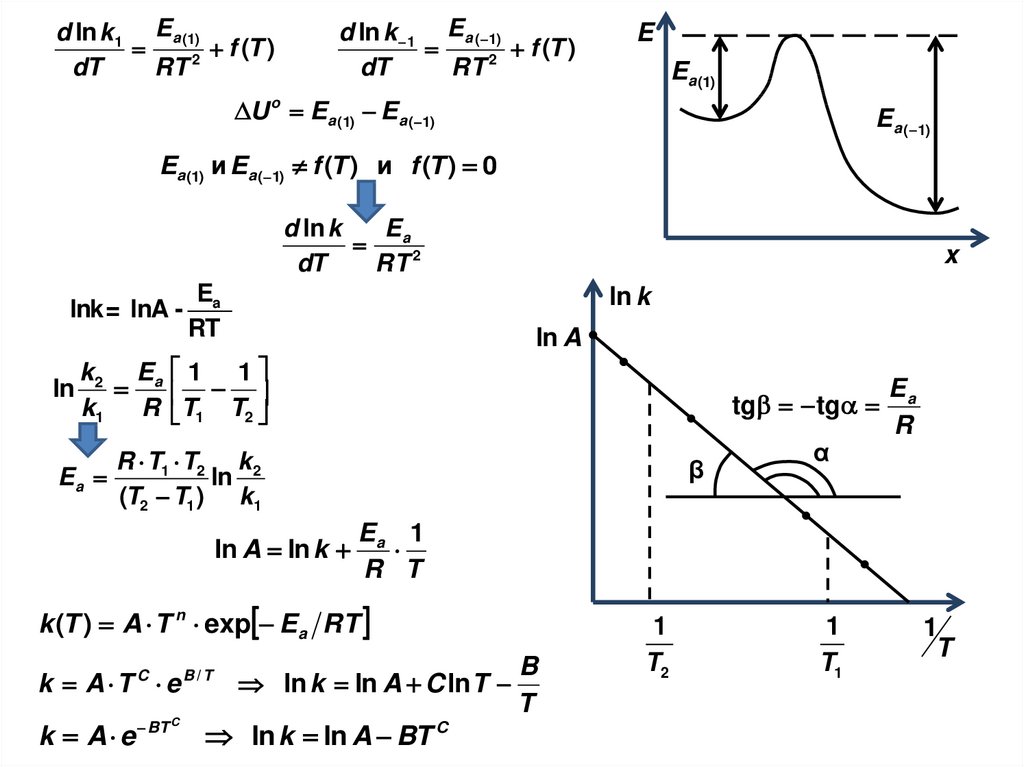
i
ΔSсм min, при x i 1
3. Принцип: min энергии — max энтропии
Δ(PV) = 0
Примем:
ΔH o = ΔU o = 8.87 к Дж/ моль
Δ Uio = 0 к Д ж / м о л ь, Δ Uno = 8.87 к Д ж / м о л ь
S io = 0 Д ж / К ×м о л ь , S no = 14.6 Д ж / К ×м о л ь
Полная энергия одного моля смеси:
Энтропия:
U m = x i× U io+ x n× U
o
n
Sm = x i× S io+ x n× S no-R x ilnx n+ x ilnx
n
114.
ТД функцияU m= x i× U io+ x n× U
o
n
1) Если энергия управляет равновесием,
то весь n-C4H10 превращается в i-C4H10
2) Если энтропия управляет процессом,
то система достигает max S при xi=0.1
3) При равновесии xi=0.89
U m- TS m
TS m
0
Компромисс
0
0.1
0.89 xi=1
Принцип Ле-Шателье:
Когда система возмущается, равновесие сдвигается в ту сторону,
которая гасит эффекты возмущения.
1.Экзотермическая реакция: тепло сдвигает в сторону исходных
реагентов
2.Увеличение числа частиц: давление сдвигает в сторону исходных
реагентов
3.Добавление исходных реагентов: увеличивает выход продуктов
115.
Температурная зависимость химического равновесия:Сдвиг равновесия (Т и Р)
1)
K p = f(T)
lnK p
R
=
T p
ΔGpo T
| ΔGpo = -RTlnK p
-
T
p
ΔGpo T
ν i μ io T
=
| ν i μ io = ΔGpo
T
T
p
p
i
μ io T
ΔH op
= – уравнение Гиббса-Гельмгольца
2
T
p
T
ΔGpo T
ΔH op
= - 2
T
T
p
ΔGpo = -RTlnKp
ΔH po
lnK p
– изобара Вант-Гоффа
=
2
T p RT
ΔHpo < 0 Kp при T (эк з о)
ΔHpo > 0
Kp при T (эндо)
116.
Примем:ΔH f(T)
o
p
lnK p = -
ΔH po
RT
+ C| K p и K' p ; T иT'|
lnKp
ΔHpo 1 1
ln
=-
K' p
R T' T
Kp
1/T
Влияние давления на химическое равновесие:
μio и ΔHpo f(P)
K x = f(P)
A = B + C (α – доля прореагировавшего А)
Начальное число молей
Число молей в равновесии
Мольные доли в равновесии
Парц. давл. в равновесии
Полное
В
С
1
0
0
1
1- α
1+ α
1- α
1+ α
α
1+ α
α
1+ α
А
1- α
×
1+ α
ρ
α
×
1+ α
ρ
α
×
1+ α
ρ
117.
Kp = K x ×α
α
×
2
α
K x = 1+ α 1+ α=
1- α
1- α2
1+ α
Kp = K x ×
νi
ρ | K
i
α2
ρ = 2×
1- α
ρ K
x
, α |
p
= Kx×
ρ
ρ | K
ρ K
x
p
= const
, α
Эффект добавления инертного газа:
Происходит ли сдвиг равновесия?
A = B + C + (n–1) моль инертного газа
Мольная доля при равновесии:
α2
Kp =
×
n+ α 1- α
V = const
A=
B=
α
α
C=
n+ α
n+ α
ρ | n , α ( при ρ = const)
PV = (n+ α)RT
α2 RT
Kp =
×
1- α V
1- α
n+ α
| α = const
P= (n+ α)×
RT
V
при любом изменении n
118.
Гетерогенное химическое равновесиеCaCO3 (тв.) = CaO (тв.) + CO2 (г)
При равновесии:
μCaCO3 = μCaO +μCO2
o
μCO2 = μCO
+RTlnρCO2
2
o
μCaO μCaO
o
μCaCO3 μCaCO
3
Приближение μ(тв. в-в) слабо зависит от P
μoCaCO3 = μoCaO +μoCO2 +RTlnρCO2 = ΔGpo = –RTlnρCO2 = –RTlnKp
Kp ρCO2
Неидеальное газовое равновесие:
μ = μo +RTlnf
ΔGpo RTlnK f
Kf
(для неидеального газа)
f L l f Mm ...
f A a f B b ...
fi
i
— коэф-т фугитивности
Pi
K f KP K γ — термодинамическая к-та равновесия,
Kγ
Ll Mm ...
Aa Bb ...
119.
Неравновесная термодинамикаКлаузиус, Кельвин 1850ые годы
Онзагер – Нобелевская премия 1968 г.
Илья Пригожин – Нобелевская премия 1977 г.
Основные понятия и определения:
dS
dqравн
T
Клаузиус:
δq'
dS
dq δq'
T
T
dq
T
– второе начало
(самопроизвольный процесс)
– второе начало
(альтернатива)
– некомпенсированная теплота
dS
Отсюда:
dS
dqравн
T
δq'
T
– второе начало
(Клаузиус)
δq' 0
– равновесные процессы
δq' 0
– неравновесные процессы
dS dвнеш S dвнутр S
равновесное поглощение
теплоты извне
dвнеш S
dqравн
T
δq'
dвнут S
T
120.
t – время неравновесного процессаλ
dвнут S
t
0 – скорость возникновения энтропии
dSU,V dвнут S 0 – изолированная система
Некомпенсированная теплота и термодинамические функции:
dq dU – pdV TdS – δq'
dU TdS – pdV – δq'
(I и II законы термодинамики)
dU TdS – pdV – δq' dUS, V – δq' 0
dH TdS – Vdp – δq' dHS,P – δq' 0
A U – TS
G H – TS
dA T, V – δq' 0
dGT,P –δq' 0
121.
Химическая кинетикаЛитература:
1. Я.И. Герасимов. Курс физической химии. II том
2. С. Бенсон. Основы химической кинетики
3. Н.М. Эммануэль, Д.Г. Кнорре. Курс химической кинетики
4. Е.Т. Денисов. Кинетика гомогенных химических реакций
5. В.М.Байрамов. Основы химической кинетики и катализа
Основные понятия химической кинетики
1. Элементарный акт (t контакта ≈ 10-13 сек)
CH3—CH3 → 2CH3
OH- + CH3Cl → CH3OH
2. Механизм химической реакции — совокупность элементарных
стадий
3. Кинетическая схема реакции — совокупность предполагаемых
элементарных стадий
4. Простая реакция
5. Сложная реакция
6. Гомогенная реакция
7. Гетерогенная реакция
122.
8. Гомолитическая реакцияH● + HR → H2 + R
9. Окислительно-восстановительная реакция
HO● + Fe+2 → OH- + Fe+3
10. Гетеролитическая реакция
OH- + R—Cl → ROH + Cl11. Мономолекулярная реакция
R—O—O—R → 2 RO
12. Бимолекулярная реакция
RН + O2 → R● + HO2
14. Реакция распада
ROOR → 2 RO
16. Реакция отрыва
A + XB → XA + B
18. Реакция рекомбинации
CH3● + CH3● → CH3CH3
13. Тримолекулярная реакция
2NO + O2 → 2 NO2
15. Реакция замещения
X + AB → XA + B
17. Реакция присоединения
Cl ● + C2H4 → C●H2CH2Cl
123.
19. Реакция диспропорционированияC2H5● + C2H5● → C2H4 + C2H5
20. Катализатор
21. Ингибитор
22. Замкнутая система
23. Открытая система
24. Молекулярность реакции
Кинетическая классификация реакций
1. Некаталитические и каталические реакции
2. Нецепные и цепные реакции
(радикальные, ионные и ион-радикальные)
3. Сложные реакции: параллельные и последовательные
4. Обратимые и необратимые реакции
124.
Скорость химической реакцииA→P
Скорость химической реакции —
количество молекул данного сорта, реагирующих
в единицу времени и в единице объема
n A n A
W
V(t t )
n A и n A — число молей одного из исходных в-в
в момент t и t
Средняя скорость
реакции
WA
V — объем системы
1 dN A
dC A
— истинная скорость
V dt
dt V
моль
моль
время объем с л
Размерность —
a1 A1 a2 A2 ... a1 A1 a2 A2 ...
dC A1
dC A2
dt
dt
1 dC A1
1 dC A2
— приведенная скорость
a1 dt
a2 dt
125.
Основной постулат химической кинетикиWA
dC A
i
dt
1
A1
k C C
2
A2
Гульберт и Вааге
1879 г.
...
αi — порядок химической реакции по компоненту Ai
i
i
— суммарный порядок реакции
k — константа скорости реакции
при CA
i
1
k = W — удельная скорость реакции
Кинетическая кривая. Кинетическое уравнение
C A0
A→P
СP → max
i
кинетическая кривая исход. реагента
кинетическая кривая продукта
СAi → 0
t
при t→∞ CP→max, CAi →0
126.
dC Ai
dt
dC
Ai
f ( CA0 , t ) — дифференциальное кинетическое уравнение
= f(C0A , t)dt — интегральное кинетическое уравнение
Сисх
Реакция останавливается
C0>0
C∞≠0
t
Спрод
dC
0
dt
Промежуточный продукт
C0≥0
dC
— меняет знак
dt
t
Формальная кинетика
1. Простые реакции (необратимые, одностадийные): W = k∙Cα
1.1. Реакции нулевого порядка α=0
1.2. Реакции первого порядка α=1
1.3. Реакции второго порядка α=2
127.
Реакции нулевого порядкаA→P
a — кол-во молей исходного А
x — кол-во молей прореагировавшего А
a – x — кол-во молей A в настоящий момент
W A P
a–x
a
d( a x ) k
( a x )0
V dt
V
d( a x )
k
dt
– дифференциальное
кинетическое уравнение
a x
d ( a x ) k t
d ( a x ) k dt
a
x k t
– интегральное уравнение
t
t½ – время полупревращения (50% А)
t = t½
x
γ =
a
a – x = 0,5∙a
t1
x = 0,5∙a
2
– степень превращения
γ = 0 при t = 0 и γ = 1 при t = ∞
k t
a
a
2k
128.
Реакция первого порядкаA→P
W k CA1
t=t
CA
t=0
nA = a
t=t
n'A = a – x
a x
V
dx
k dt
a x
d( a x ) k
dx
(a x )
k (a x )
V dt
V
dt
dx
a x k dt
ln(a x ) k t C
1 C0
k ln
t
C
x – прореагировало
t=0
ln a C
x=0
Размерность
k = t-1 (сек-1, мин-1…)
1
a
k ln
t
a x
a
e k t
a x
t½→a/2 = x
a / 2 a(1 e k t ) ,
2 e k t 1 ,
a
C0
V
a x
C
V
a x a e k t
1 2 2 e k t ,
e k t 2 ,
k t ln 2
x a(1 e k t )
t1
2
ln 2
k
129.
Реакция второго порядкаR–COOR’ + NaOH → RCOONa + R’OH
A+B→C+D+…
и
t=0
nA = a
t=t
n'A = a – x и
nB = b
n'B = b – x
x – прореагировало A и B
Объем системы = V
W k CA CB
CA
a x
V
CB
b x
V
d (a x )
a x b x
dx k
k
(a x ) ( b x )
V dt
V
V
dt V
V = const
k
k
V
dx
k (a x ) ( b x )
dt
dx
(a x ) (b x ) k dt
1 1
b( a x )
k
ln
t a b
a (b x )
130.
Размерность[A] = [B] = a
[k] = время-1∙моль-1
[k’]= время-1∙моль-1 ∙объем
dx
dx
k (a x ) 2
k dt
2
dt
(a x )
1
x
k
t a (a x )
t = t½
x = a/2
k
1
t1
2
a
1 1
2
a
a (a 2 ) t 1 a
t1
2
1
k a
2
Реакция α-порядка
A+B+…→C+D+…
[A] = [B] = … = a
V = const
dx
= k× ( a- x) α,
dt
1 1 a 1 (a x ) 1
k
1
t 1 a (a x ) 1
t1
2
1 1 2 1 1
k 1 a 1
131.
Определение констант скоростейln(a–x)
ln a ln(a x ) k t
lna
I – порядок
tgα = –k
1
a
k ln
t
a x
α
1
CA
t
II – порядок
α
1
C A0
ln
a b
a x
C A0
CA
V V
V
x
CA0 CA
1
1
k t
0
0
(a x ) a C A C A C A C A
tgα = k
t
a x
b x
II – порядок a ≠ b
b
ln
a
α
1
x
k
t a (a x)
tgα = k∙(a–b)
k t
t
1
b( a x )
ln
a b
a (b x )
132.
Определение порядка реакцииМетоды: 1) Проб и ошибок
2) Метод Вант-Гоффа
3) Метод Раковского
4) Метод Оствальда-Нойеса
1)
V k [ A] 1 [B ] 2
( 0) CA CA0 k A t
( 1) ln CA0 ln CA k A t
1
1
( 2)
kA t
C A C A0
N
1.1 Расчетный
Δk→min
CA
k
t
k
k
(α=0) (α=1) (α=2)
1
2
3
1.2 Графический
CA
lnCA
α=0
t
1/CA
α=1
t
α=2
t
133.
2) Метод Вант-ГоффаCA
W1=tgβ1
C1
lgWA
W2=tgβ2
C2
ΔlgWA
t1
t2
t
dC1
dC 2
k C1 ;
k C2
dt
dt
lg W1 lg W2
W1 C1
| lg
lg C1 lg C2
W2 C2
tgβ = α
β
ΔlgCA
lgCA
3) Метод Раковского (по t½)
a
t 1 f (a )
2
2
2k
ln 2
3.2. 1 t 1
t 1 f (a )
2
2
k
2 1 1
3.3. 2,... n (a b ... ) t 1
2
k ( 1) a 1
3.1. 0
t1
(t 1 )
a1
2
(t 1 )
a2
2
1
134.
lgt½β
1
tgβ = α-1
lg(t 1 ) lg(t 1 )
2
lg a2 lg a1
lga
4) Метод Оствальда–Нойеса
Определяется частный порядок по реагенту
dC1
k C1 1 C2 2 C3 3
dt
dC1
k C1 1
dt
C1 C2 и C3
C2 C1 и C3
-
dC2
= k × Cα2 2
dt
C3 C1 и C2
dC3
= k × C3α3
dt
2
135.
Сложные реакции4 типа сложных реакций
1. Обратимая реакция
A
2. Параллельная реакция A
B
B
C
3. Последовательная реакция A
B
C
4. Цепная реакция
Принцип независимости:
Если в системе одновременно протекает несколько реакций,
то каждая из них независима от остальных и скорость ее
пропорциональна концентрации реагирующих веществ.
136.
1. Обратимая реакция первого порядкаA
k1
k2
B
a и b – кол-во молей A и B при t = t 0
x – кол-во молей A прореагировавших к моменту t
d (a x )
k 1 (a x ) k 2 (b x )
dt
k1 a k1 x k2 b k2 x k1 a k2 b x (k1 k2 )
k 1 a k 2 b
x
(k1 k 2 )
k 1 k 2
k1 a k 2 b
L
k1 k 2
dx
(k1 k 2 ) (L x )
dt
dx
(L x ) (k1 k2 ) dt
ln
L
t (k 1 k 2 )
L x
1
L
k1 k 2 ln
t L x
137.
KPk1
k2
L
KP a b
1 KP
L
k1 a k 2 b
k1 k 2
dx
C
0 в момент равновесия
x
dt
∞
x∞ – кол-во молей А в равновесии
k1 (a x ) k2 (b x ) 0
k1 b x
KP
k2 a x
dx
k 1 (a x )
dt
x∞
t
k1 a k2 x k2 b k2 x
dx
k 2 (b x )
dt
k1 a k2 b (k1 k2 ) x
k1 a k 2 b
x
L
k1 k 2
t (k1 k 2 ) ln
b
a
x
x x
1
L
k1 k 2 ln
t
L x
ln
x
x x
α
tgα = k2 + k1
t
138.
2. Обратимая реакция второго порядкаA+B
C+D
d (a x )
k1 CA CB k 2 CC CD
V dt
a – исходное кол-во молей вещества A (а = b);
x – число молей вещества А, прореагировавшего к моменту t;
V – объем системы;
k’1 и k’2 – константы скоростей прямой и обратной реакции;
Ci – концентрация реагирующего вещества к моменту t.
dx
k1 C A CB k 2 CC CD
V dt
dx
(a x ) 2
x2
k1
k2 2
2
V dt
V
V
1
V
dx
k 1 (a x ) 2 k 2 x 2
dt
k1
2
dx
k1 a
k1 a 2
(k 1 k 2 ) x 2
x
dt
k
k
k1 k 2
1
2
2 a K
a2 K
x
x
0
K 1
K 1
k
K 1
k2
2
dx
(k1 k 2 ) (m1 x )(m2 x )
dt
m1,2
a(K K )
K 1
1
1
m (m x )
k1 k 2
ln 2 1
t m1 m2
m1(m2 x )
k1
k
и k2 2
V
V
139.
Параллельные реакцииdx 1
1 B
k 1 (a x )
A
dt
2 C
dx 2
k 2 (a x )
I порядок
dt
x1 и x2 – число молей вещества B и C к моменту t;
x1 + x2 = x – общее число молей A прореагировавших к моменту t;
k’1 и k’2 – константы скоростей реакций 1 и 2.
dx
k 1 (a x ) k 2 (a x )
dt
dx dx 1 dx 2
dt
dt
dt
dx
(k1 k 2 )(a x )
dt
1
a
k1 k 2 ln
t a x
dx 1 k1
dx 2 k 2
x
dx
0
x
1
k 2 dx 2 k1
0
x 1 k1
x 2 k2
II порядок
dx
(k1 k 2 )(a x )(b x )
dt
k1 + k2 =
1
b(a- x)
ln
(a- b)t a(b- x)
140.
Последовательные реакции1
2
A
B
C
мономолекулярные реакции
t = 0 a – исходное кол-во молей вещества A (а = b); x – число молей вещества А,
прореагировавшего к моменту t;
t = t a–x – число молей вещества А, x–y – число молей вещества B;
y – число молей С.
k1 и k2 – константы скоростей реакций 1 и 2.
Для A:
Для B:
dx
k 1 (a x )
dt
a x a e k1 t
a
d (x y )
k 1 (a x ) k 2 ( x y )
dt
y
d (x y )
k1 a e k1 t k 2 ( x y )
dt
x–y
d (x y )
k 2 ( x y ) k1 a e k1 t
dt
k1
k t
k t
x y a
(e 1 e 2 )
k 2 k1
x a(1 e
k2
k1
-k t
-k t
y= a 1.e 1 +
.e 2
k2 - k1
k2 - k1
a–x
п.и.
k1 t
)
tmax
t
п.и. – период индукции
в tmax
d (x y )
0
dt
141.
d (x y )k1
k t
k t
a
( k1 e 1 k 2 e 2 ) 0
dt
k 2 k1
k1 e
k1tmax
k2 e
ln k1 ln k 2
k1 k 2
ln r
(r 1) k1
k2tmax
k2
r
k1
t max
t max
ln
a
k1
0
k2 -k1
ln k1 k1 tmax ln k2 k2 tmax
k2 r k1
-r
a
(x - y) max =
(e
1- r
lnr
r-1
-
-e
lnr
r-1
)
x - y= a
k1
-k t
-k t
(e 1 - e 2 )
k2 -k1
Максимальная концетрация В зависит от соотношения констант
k 2 t
k1 t
k1
-k t
-k t
(e 1 - e 2 )
k2 -k1
e
1. k1 k2 e
a k1
x y
e k1 t
a e k1 t a x
k 2 k1
(a x ) k 1
x y
k1
x y
const
k 2 k1
a x k 2 k1
Переходное состояние
2.
k1 k2
x - y= a
x - y k1
=
a- x k 2
Вековое равновесие
k1
k2
-k t
-k t
k1 k2 y a(1 e k1 t )
y= a 1× e 1+
× e 2
k2 - k1
1я стадия лимитирующая
k2 - k1
142.
k2 k1Метод квазистационарных концентраций (Боденштейн)
x y k1 2
k
x y (a x ) 1
k2 ( x y ) k1(a x )
a x k 2 1
k2
d (x y )
0
dt
1
2 –
HO–CH2–CH2–Cl + OH
k2 ( x y ) k1(a x ) 0
3
4
CH2–CH2 + H2O + Cl–
O
k1
–
1. HO–CH2–CH2–Cl + OH
2. –O–CH2–CH2–Cl
k2
k–1
–
O–CH2–CH2–Cl + H2O
[X] – промежуточное в-во
CH2–CH2 + Cl–
O
(V2)
(V1)
(V–1)
dC X
k1 C1 C2 k 1 C X C4 k 2 C X 0
dt
CX
V2
k1 C1 C2
k 1 C4 k 2
dC 3
k2 CX
dt
dC 3 k1 C1 C2 k 2
– скорость образования CH2–CH2
dt
k 1 C4 k 2
O
143.
V V1 V 1 V2С4 const (в воде)
V k эфф C1 C2
k1 k 2 C1 C2
k 1 C4 k 2
k эфф
квазистатический режим
V1 V 1 V2 0
k1 k 2
k 1 C4 k 2
Цепные реакции
– реакции со сложными превращениями исходных веществ в продукты,
осуществляемые при регулярном чередовании нескольких реакций с
участием активных центров.
Активный центр: атом или свободный радикал.
Три основные стадии:
1. Стадия зарождения C2H6
2CH3
2. Стадия развития (продолжение и разветвление цепи)
C2H4 + Cl
C2H4Cl
R● + O2
ROO
CH4 + Cl
CH3 + HCl
CH3OO
● CH
H● + O2
● OH
◄ развлетвление
+ ●O
3. Обрыв цепи
H● + H
H2
● CH
3+
●CH
3
C2H6
2OOH
продолжение
144.
Кинетика цепных процессовd [COCl2 ]
CO + Cl2
COCl2
k [CO][ Cl2 ]3 / 2 – эксперимент
dt
Предполагаемый механизм:
Cl2 + M
k1
CO + Cl
k2
2Cl● + M
М – активирующая частица
COCl
k3
COCl2 + Cl
COCl● + Cl2
k4
COCl
CO + Cl
k5
Cl + Cl + M
Cl2 + M
d [COCl2 ]
k 3 [COCl ][ Cl2 ]
dt
d[ Cl• ]
dt
= k1 × [ Cl2 ] [ М ] + k3 × [ CO Cl• ] [ Cl 2 ] + k4 × [ CO Cl• ] - k 2 × [ Cl• ] [ CO ] - k5 × [ Cl• ] 2 [ M]
d [COCl ]
k 2 [Cl ][ CO] k 3 [COCl ][ Cl2 ] k 4 [COCl ]
dt
Принцип стационарных концентраций (Боденштейн)
d [Cl ]
0
dt
d [COCl ]
0
dt
145.
Кинетика фотохимических реакцийА + hν → A*
А*
А*
A*
A*
A*
(Cl2 + hν → 2Cl●*)
→ B – реакция внутримолекулярного превращения
→ B● + D● – реакция распада
+ B → AB – реакция взаимодействия
+M→A+M
дезактивация
→ A + hν
Законы фотохимии
З-н Гротгуса-Дрепера: только поглощаемое средой световое
излучение может вызвать ее химическое изменение.
З-н Эйнштейна: каждый поглощенный квант света в первичном акте
способен активировать только одну молекулу.
Вант-Гофф: количество химически измененного вещества
пропорциональна количеству поглощенной световой энергии.
Закон Бугера-Ламберта–Бера: Jt = J0∙e–ε∙c∙l
Jt – интенсивность светового потока после прохождения;
J0 – начальная интенсивность светового потока;
ε – коэффициент поглощения; с – концентрация вещества;
l – толщина кюветы
146.
Ja = J0 – Jt = J0 – J0∙e–ε∙c∙l = J0(1 – e–ε∙c∙l )dC
k J 0 (1 e cl )
dt
Ja
1. c l 1
dc
k J0
dt
2. c l 1 e
cl
Вант-Гофф
(нулевой порядок)
1 c l
-
Первичный квантовый выход =
(γ1)
Полный квантовый выход =
dc
= k c,
dt
(первый порядок)
Число прореаг. молекул в первичной р-ции
Число квантов (поглощенных)
Число квантов (поглощенных)
γ1 ≤ 1
γ ≥ 1 или γ ≤ 1
γ = 105
(COOH)2 → CO + CO2 + H2O
k= k l J0
Число образовавшихся молекул продукта
Первичная реакция: А + hν → A*
H2 + Cl2 → 2HCl
- поглощенная энергия
γ = 10-3
147.
Элементарный акт фотохимической реакцииАB + hν → AB*
→ А + B – диссоциация
→ BA – изомеризация
+D
AB* →
→ AD + B – хим. реакция с изменением состава
S1 → S2
T1 → S1
→ AB + hν – излучение (флуоресценция или фосфоресценция)
→ AВ – внутримолекулярный переход энергии
→ AB + CD* – дезактивация при соударении или на стенке
CD*
Кинетическая схема Штерна-Фольмера (упрощенная схема)
k
1
Поглощение АB + hν →
AB*
k
Флуоресценция АB* →2 AB + hν
k
v1 = Ia (интенсивность)
v2 = Iфл=k2∙[AB*]
3
Дезактивация AB* + AB →
AB + AB
k
4
Диссоциация АB* →
A+B
v3 = k3∙[AB*][AB]
v4 = k4∙[AB*]
Ia – интенсивность (число квантов, поглощенных в единицу t (время) и
в единице V (объем))
Iфл – число квантов, испускаемых флуоресценцией в единицу t (время)
из единицы V (объем)
148.
d[ AB* ]= I a - k 2 × [ AB* ] - k3 × [ AB* ] [ AB] - k 4 × [ AB* ] = 0
dt
(принцип квазистационарных концентраций)
Ia
[ AB*]
v 4 k4 [ AB*]
k 2 k 3 [ AB ] k 4
Ia
Ia
d [A]
k4
dt
k 2 k 3 [ AB ] k 4 1 k 2 k 3 [ AB ]
k4 k4
v4
1
1
Первичный квантовый выход 1 v
k
k
1
1 2 3 [ AB ]
k4 k4
1
I a Iфл k 3 [ AB*][ AB] k 4 [ AB*] Iфл k 2 [ AB*]
Iфл
Ia
k
I
k
1 4 3 [ AB] a тушение
Iфл
k2 k2
Iфл
Ia
Ia
v4
v
1
4
1
k
k
Iфл
v1
1 2 3 [ AB ]
k3
k4 k4
k
v4
α
tg
3
k2
k4
k
1
k
1 2 3 [ AB ]
1
k4 k4
k4
k2
[AB]
149.
11
α
tg
k
1
k2
f ( AB)
1 и 3
1
k4
k4
k3
k4
k2 – можно измерить независимым путем
k
1 2
k4
[AB]
Зависимость скорости реакции от температуры
kT2
k
T 10 2 4
kT
Аррениус k A e
kT1
EA
RT
A и E A f (T )
1 1
k1(T1 )
exp C
k 2 (T2 )
T1 T2
Вант-Гофф
T2 T1
10
d ln KC U o
dT
RT 2
C
Ea
R
(изохора Вант-Гоффа)
KC – константа равновесия реакции
k1
dlnk1 dlnk-1 ΔU o
KC =
=
k-1
dT
dT
RT 2
150.
d ln k1 E a (1)f (T )
dT
RT 2
d ln k 1 E a ( 1)
f (T )
dT
RT 2
E
Ea (1)
U o Ea (1) Ea ( 1)
E a ( 1)
Ea (1) и Ea ( 1) f (T ) и f (T ) 0
E
d ln k
a2
dT
RT
lnk= lnA -
ln
Ea
RT
x
ln k
ln A
k2 Ea 1 1
k1 R T1 T2
tg tg
R T1 T2 k 2
Ea
ln
(T2 T1 ) k1
ln A ln k
β
Ea 1
R T
k (T ) A T n exp E a RT
k A T C e B / T ln k ln A C lnT
k A e BT
C
α
Ea
R
ln k ln A BT C
B
T
1
T2
1
T1
1
T
151.
Скорость реакций в открытых системахРеактор идеального смешения
С0,A υ
Сi,A υ
A → P (продукты)
С0,A – концентрация А на входе (моль/м3);
Сi,A – концентрация А на выходе (моль/м3);
υ – скорость входа и выхода потока (м3/сек);
V – объем реактора (м3); nA – количество молей А
rA – скорость превращения А→P
dn A
1
dn A
dC A
C0,A Ci ,A rA V |
dt
V
V dt
dt
dC A
(C0,A Ci ,A ) rA
dt
V
dC A
dC A
rA (C0,A Ci ,A )
?
V
dt
dt
Стационарный режим:
× C0, A = × Ci, A + rA × V
dC A
0 rA (C0,A Ci ,A(СТ ) )
dt
V
Сi,A(СТ) – концентрация А при
стационарном режиме.
152.
Кинетика реакций в реакторе идеального смешенияA → P (продукты) (мономолекулярная)
r
dC A
(C0,A Ci ,A )
V
dt
r k CA
dC A
(C0,A Ci ,A ) k C A 0
dt
V
t 0
t t
интегрирование
CA =
× 1- e
+ k× V
C0, A ×
t
C
CT
A
+ k× V . t
V
C0,A
k V
Стационарное состояние (t = )
1
Пусть k 1 сек ,
t 10 сек
e
+ k× V
V
достигается быстро
0.1 м3 / сек
V 1м3 ,
-
t
0
153.
В промышленности:υ∙ С0
υ∙ Сi
Ci 0,001 C0,1
на выходе
1
→
A1 ← A2
–1
υ
rA = (C0,A - C i,A( СТ ) )
V
После установления стационарного режима
k1 C1CT k 1 C2CT
k1 C1CT k 1 C2CT
(C0,1 C1CT )
V
C2CT
V
C0,1 C1CT C2CT
если
A2 – не вводится в реактор (С0,2 = 0)
C2
C1
CT
k1
k 1 V
C2
при k 1 V
C1
C2
– не зависит от времени
C1
C2
k1
=
C1 k-1 +
V
KC =
k1
k-1
154.
Реактор идеального вытесненияdl
C0,A
S
(CA dCA )
CA
CL , A
L
Sdl – элемент объема, υ – объемная скорость движения реакц. смеси (м3/с)
rA – скорость превращения А
Стационарный режим:
CA (CA dCA ) rA Sdl
dC A rA Sdl
rA
dC A
S dl
dC A
– изменяется концентрация
dl
155.
Кинетика реакций в реакторе идеального вытесненияk1
A1 P
dC A1
k1 C A
1
S dl
rA k 1 C A
l 0
CA C0,A
l l
CA Cl ,A
1
1
1
1
rA =
×
S
1
ln
C0,A
1
Cl , A
1
Cl ,A C0,A e
1
k S l
1
k1 S l
– концентрация Ai вдоль реактора
1
l L – длина реактора
CL,A C0,A e
1
k S L
1
– концентрация на выходе
1
S L V – объем реактора
CL,A C0,A e
1
1
k V
1
υ – скорость подачи реагента
dC A
dl
156.
КАТАЛИЗИзменение скорости химических реакций под влиянием веществ —
катализаторов, многократно вступающих в промежуточное химическое
взаимодействие с участниками реакции, восстанавливающих после
каждого цикла свой химический состав.
1. Положительный и отрицательный (ингибирование)
2. Гомогенный, гетерогенный и ферментативный (микрогетерогенный)
3. Гомолитический и гетеролитический
Основные типы механизмов катализа
1. «Переносный» катализ (перенос атома, молекулы, электрона)
SO2 + V2O5 → SO3 + V2O4
2SO2 + O2 → 2SO3
V2O4 + ½O2 → V2O5
2. «Активационный» катализ
H—H
Pt Pt
H H
Pt Pt
H2 → 2H
ΔpH = 430 кдж/моль
ΔH ≈ 250 кдж/моль, для H∙ ∙ ∙Pt
3. «Координационный» катализ: С2H4 + ½O2 → CH3CHO (Cu2+, Pd2+)
PdCl2 + H2O + C2H4 → Pd0 + CH3CHO + HCl
2Cu2+ + Pd0 → 2Cu+ + Pd2+
2CuCl + 2HCl + ½O2 → 2CuCl2 + H2O
157.
Cl–Cl–
–
OH
Pd+2
1-–
Cl–
Cl–
C2H4
Pd0
[ ]
2–
[ ]
CH3CHO + H+
Избирательность. Активный центр.
Cu
C2H5OH
Al2O3
Al2O3
Zn, Cr2O3
CH3CHO + H2
T = 473 – 523 K
(C2H5)2O + H2O T = 523 K
C2H4 + H2O
T = 623 K
CH2=CH–CH=CH2 + H2 + H2O
T = 673 – 723 K
Активный центр – группа атомов или ионов в кристаллической решетке
на поверхности
– участок в молекуле белка
Эффективность катализатора. Активность.
Для гомогенного катализа:
nk
Cкат
– число оборотов реакции: кол-во молекул превращающихся
за единицу времени на одном активном центре (υ – скорость).
Для гетерогенного катализа:
активность
a
A кат
– удельная активность; А – кол-во реагента или продукта
S
S
с единицы массы катализатора
площадь поверхности катализатора
158.
Интегральная избирательность – степень превращения реагента в целевойпродукт → f(t, состав реакционной смеси, конечная степень превращения)
скорость образования целевого продукта
(цп )
– дифференциальная селективность
i
скорость по всем направлениям
Теория промежуточных соединений в катализе (Шпитальский)
1. Катализатор образует с исходным веществом неустойчивое промежуточное
соединение (ПС)
2. Образование промежуточного соединения протекает обратимо и достаточно
быстро
3. Неустойчивое промежуточное соединение относительно медленно
претерпевает дальнейшие превращения в направлении образования
конечного продукта и выделения катализатора
4. Общая скорость каталитической реакции пропорциональна концентрации
промежуточного соединения, но не реагента
5. Один и тот же катализатор может взаимодействовать с реагентом, образуя
одновременно несколько промежуточных соединений
159.
Механизмы каталитических явлений в рамкахтеории промежуточных соединений
а) реакции с одним реагентом:
k1
k2
A + Кат →
← АКат →
Р + Кат
k-1
A→P
б) реакции с двумя реагентами:
A+B→P
1
2
A + Кат → АКат; Aкат + B → P + Кат
в) реакции с двумя реагентами с образованием двух продуктов (I):
A + B → P1 + P2
1
2
A + Кат → P1 + Кат∙Х; Кат∙Х + B → P2 + Кат
г) реакции с двумя реагентами с образованием двух продуктов (II):
A + B → P1 + P2
k1
A + B + Кат →
← AКатВ и АКатВ
k-1
k
→2 Р1 + Р2 + Кат
Термодинамические аспекты катализа
ΔrG > 0 не идет в прямом направлении,
катализатор ускоряет обратную реакцию
Δ rG < 0
ΔGpo RTlnK f
Катализатор одинаково ускоряет скорости прямой и обратной реакции,
но не влияет на K (не путать с Kc)!
160.
Определения:AH
BOH
Кислотно-основной катализ
A– + H+
B+ + OH–
Аррениус
HB+ +
HA + B
р-ль H2O
A–
Бренстед
кислота основание кислота основание
сопряженные
A
+ : B
Льюис
A– B+
FeCl3, AlCl3, BF3 – примеры кислот
NH3, N2H4, C5H5N – примеры оснований
Типы кислотно-основного катализа
Специфический кислотный катализ
H+
+ H2O
H3
O+
S + H3
CH3C–OC2H5 + H3O+
OH
CH3C–OH
+
H O C2H5
–H2O
+H2O
k-1
SH+
CH3C–OC2H5
+H2O
–H2O
–C2H5OH
+C2H5OH
CH3C
OH
+H2O
–H2O
k2
P + H3O+
медленно
OH
+
CH3C–OH2
+
O H
+ H2O
быстро
+
O H
O
k1
O+
OC 2H5
O
+ H3O+
CH3C
OH
161.
Общий кислотный катализS + HA
k1
k2
SH+ + A–
медленно
CH3—CH
HA – кислота Бренстеда
быстро
CH—OR + AH
CH2
P + HA
медленно
+
O R + H2O
CH3—CH
+
O R + A–
CH3—CH—OR
+
OH
H
CH3—CH—OR
CH3—CH—O+R
OH
H
OH
+
CH3—CH—O+R
OH
–ROH
H
CH3CH=O+H
H
+H2O
–H2O
CH3CH=O + H3O+
Электрофильный катализ
A—B: +
E+
k1
k–1
+
A—B—E
N–
+
+ A—B—E
k2
..
N–A + B–E
162.
Реакция Фриделя-Крафтса[C2H5]+[AlCl4]–
C2H5Cl + AlCl3
C2H5
+ [C2H5]+[AlCl4]–
+ HCl + AlCl3
Специфический основный катализ
SH +
k1
OH–
k–1
S–
+ H2O
S* +
быстро
CH3CHO + OH–
CH2CHO + CH3CHO
OH–
k2
P + OH–
медленно
CH2CHO + H2O
CH3–CH–CH2–CHO
-
быстро
медленно
O
CH3–CH–CH2–CHO
-
O
CH3–CH–CH2–CHO + OH–
OH
163.
Общий основный катализSH + B
k1
S–
медленно
+
BH+
S* + B
[CH2CH=O]– + [BH]+
CH3CHO + B
–
[CH2CH=O] + CH3CHO
CH3–CH–CH2–CHO
k2
P+B
быстро
[BH]+
-
O
CH3–CH–CH2–CHO + B
OH
Нуклеофильный катализ
2C6H5CHO
CN–, H2O
OH
C6H5CO–C–C6H5
-
C6H5CHO + CN–
OH
C6H5C
–
CN
O
C6H5–C–H
CN
+ C6H5CHO
-
O OH
C6H5–C–CH–C6H5
CN
OH
C6H5C–
CN
OH O
C6H5–C–CH–C6H5
CN
OH
C6H5–CO–CH–C6H5 + CN–
164.
Кинетика реакций кислотно-основного катализаk1
+
1. SH + H3O
HSH+ + H2O – кислотный специфический катализ
k2
–
2. SH + ОН
S– + H2O –основной специфический катализ
k3
3. SH + H2O
HSH+ + OH– – кислотный общий катализ
k4
4. SH + H2O
S– + H3O+ –основной общий катализ
SH 1 2 3 4
d [ SH]
k каж [ SH] (k 3 k 4 )[H2O][ SH] k1[H3O ][ SH] k 2 [OH – ][ SH]
dt
k
Пусть: k 3 k 4 k 0
константа скорости
взаимодействия с водой
(«некаталитическая»)
SH
4
k1 [H3O ] kH [H ]
k H – константа скорости катализа ионами H+
k2 [OH– ] kOH– [OH– ]
k OH – – константа скорости катализа ионами OH–
kкаж k0 kH [H ] kOH– [OH– ]
k каж k0 kH [H ]
–
kOH
[H ]
[H ][OH– ] KH2O
– k OH– KH2O
k OH
165.
lgkкажe
a
f
b
d
c
H+
OH–
H2O
pH
a – три области (H+, H2O, OH–)
b – нет области катализа H2O
в точке min kH [H ] kOH– [OH– ]
c – при рН>7 только OH–
d – только OH–
e – H+ при pH<7
f – только H+
[H ]min
1 kOH– KH2O
pH lg
2
kH
k OH– KH2O
k H
1/ 2
166.
CH3COCH3 + X2(галоген) → CH3COCH2X + HXКислая среда: k [CH3COCH3 ][H3O ]
+
CH3COCH3 + H3O
KP
k1
k–1
CH3–C–CH3 + H2O
[ YH ]
[CH3COCH3 ][H3O ]
1. YH+ + OH–
k2
YH+
+
HO
CH3–C=CH2 + H2O
OH
k3
2. YH+ + H2O
CH3–C=CH2 + H3O+
OH
3. YH+ + A–
k4
CH3–C=CH2 + HA
OH
d [ енол]
k 2 [ YH ][ OH – ] k 3 [ YH ] k 4 [ YH ][ A – ]
dt
[ YH ]
Kp
[CH3COCH3 ][H3O ]
d [ енол]
K p [CH3COCH3 ][H3O ] k 2 [OH ] k 3 k 4 [ A – ] ( )
dt
*
167.
KH2O [H3O ][OH – ]K a [HA] [H3O ][A – ]
d [ енол]
K p [CH3COCH3 ] k 2KH2O k 3 [H3O ] k 4K a [HA]
dt
( )
*
CH3–C=CH2 + X2
k5
быстро
CH3COCH2X + HX
OH
d[ CH3 COCH 2 X]
= K р[ CH3 COCH3 ] k0 + kH+ [ H+ ]+ kHA [ HA]
dt
k0 = k2 K p KH2 O ( не к а т а лит иче с к а я ча с т ь); kH+ = k3 Kp (с пе ц. );
kHA = k4 Kp K a(общ ий)
kкаж k0 kH [H3O ]
Если [HA] ≈ 0
k каж
α
tg kH
k0
[H3O+]
168.
Реакция катализируется основаниямиk6
1. CH3COCH3 + OH–
CH3COCH2 + H2O
k7
2. CH3COCH3 + H2O
CH3COCH2 + H3O+
k8
3. CH3COCH3 + B–
CH3COCH2 + BH
k9
CH3COCH3 + X2
CH2
C
– O
быстро
CH3COCH2X + X–
[CH3COCH3 ] k 7 k 6 [OH – ] k 8 B –
Кислотно-основной катализ (H+ и OH–)
d [CH3COCH2 X ]
[CH3COCH3 ] k 0 kH H3O k OH– [OH – ]
dt
k каж k0
k0 kH
k H O k
H O k K
H
OH –
3
3
OH
–
lgkкаж
[OH ]
–
H2O H3O
1
tgα=–1
α
α
tgα=1
pH
169.
Уравнение Бренстеда (общий кислотно-основной катализ)kHA CA K A
kB CB KB
kHA и kB – константы скорости
CA, CB, α и β – константы (α и β < 1)
гомологический ряд
KA и KB – константы ионизации кислот и оснований
ln kHA lnCA ln KA;
ln kB lnCB ln KB
Для серии
Ki
ki Ki
ki
ln ln
k1 K1
k1
K1
G
K P exp
RT
G
kБT
kБT
k æ
K æ
exp
h
h
RT
Для растворов: G F
F const F
ln kHA lnCA ln KA
0 1
S и SP – константы
Ea const HP
F U T S
170.
Ферментативный катализФерменты — биологические катализаторы белковой природы,
ускоряющие реакции в живых системах
Основное отличие — специфичность (4 типа):
1. Абсолютная специфичность
2. Абсолютная групповая специфичность
3. Относительно-групповая специфичность
4. Стереохимическая специфичность
Кинетика ферментативных реакций с одним субстратом
S – субстрат; P – продукт; E – фермент
S
P
d [P]
k 2 [ES]
dt
E+S
k1
k–1
ES
k2
P+E
d [ES]
k1[ S ][E ] k 1[ES] k 2 [ES]
dt
[E]0 = [E] + [ES] и [S]0 = [S] + [ES] + [P]
[E]0 и [S]0 – начальные концентрации фермента и субстрата
[E] и [S] – текущие концентрации фермента и субстрата
[ES] и [P] – концентрации промежуточного соединения и продукта
171.
k ækБT A B
K
k0 A B – уравнение Бренстеда–Бьеррума
h
AB
AB
k0 – константа скорости при бесконечном разбавлении (γ=1)
Дебай–Хюккель
A ZA
k k0 A B
AB
lg z A zB h I → средний коэф-т активности
B ZB
AB Z A ZB
k
A B
k0
AB
A B
k
lg lg
k0
AB
lg z A zB h I
h Z A2 Z B2 Z A Z B I 2h Z A Z B I
k
lg 1.02 Z A Z B I
k0
k
lg
k0
2
1.826 106
h
( T ) 3 / 2
Z A ZB 0
Z A ZB 0
I
Z A ZB 0
при 25ᵒС → h = 0.51 (H2O)
172.
Условия:1. t→0 P →0
[S]0 = [S] + [ES]
2. [S]0 >> [E]0
[S]0 ≈ [S]
3. [ES] – стационарна
d[ES]/dt = 0
k1[S]0 [E]0 [ES] k 1[ES] k2 [ES] 0
k1[S]0 [E]0 k1[S]0 [ES] k 1[ES] k2 [ES] 0
[ES]
KM
0
k1[E]0 [ S]0
k1[ S]0 k1 k 2
k 1 k 2
k1
1
k1
[ES]
[E]0 [ S]0
[ S]0 K M
– константа Михаэлиса
k 2 [E]0 [ S]0
[ S]0 K M
1. [S]0 << KM
2. [S]0 >> KM
0 k2 [E]0 max
0 k 2 [ES]
0
k 2 [E]0
[ S]0
KM
0 k 2 [E]0
1 порядок по S и E
0 порядок по S, 1 порядок по E
[ES] → [ES] max (весь фермент в ES)
173.
0max [ S ]0
[ S ]0 K M
при 0
Уравнение Михаэлиса-Ментен
max
2
K M [ S]0 – физический смысл
KM
при k–1 >> k2
KM f (T ,pH)
k 1 k 2 k 1
K S – субстратная константа,
k1
k1
константа диссоциации
комплекса ES
KM
при k2 >> k–1
k2
k2
k1
K M KS
max
моль л
мин 1 – число оборотов фермента
[E ]0 ( л мин) моль
max
0
0 порядок
max
2
I порядок
KM
[ S]0
174.
Способы определения кинетических параметровферментативных реакций
2
Координаты Лайнуивера-Берка
1
0
max
1
0
K
tg M
max
max
1
KM
1
KM
0
1
1
K
1
M
0 max max [ S ]0
Координаты Вульфа-Хейнса
[ S ]0
0
KM
max
KM
1
[ S ]0
0
tg
max [ S ]0
K M [ S ]0
[ S ]0
1
max
[ S ]0
K
1
M
[ S ]0
0
max max
[ S]0
175.
0Коордианты Эди-Хофсти
1
1
K
1
M
0 max max [ S ]0
tg KM
max
0 max
tg KM
0
max
0 max K M
0
[ S ]0
0 [ S]0
KM
Ингибирование ферментативных реакций
Типы ингибирования:
1) Ингибитор (I) конкурирует за активный центр с субстратом
2) Ингибитор не конкурирует с субстратом за активный центр.
(Уменьшение активности фермента за счет комплексов EI и ESI)
Конкурентное ингибирование
E+S
k1
k–1
ES
k2
P+E
1. t→0 P →0
[S]0 = [S] + [ES]
2. [S]0 >> [E]0
[S]0 ≈ [S]
3. [ES] – стационарна
d[ES]/dt = 0
неактивный
комплекс
[E][I] константа
KI
[EI] ингибирования
E+I
EI
176.
[I]0 >> [E]00
[I] ≈ [I]0 – начальная концентрация I
d [P]
k 2 [ES]
dt
[E]0 = [E] + [ES] + [EI]
[I]0
[E]0 [ES] [E] 1
KI
[EI]
[E][I]
KI
d [ES]
k1[E ][ S ] k 1[ES] k 2 [ES] 0 [S]0 = [S]
dt
k [E ][ S ]0 [E ][ S ]0
[ES] 1
k 1 k 2
KM
k k
[ES]K M
[I]
[ES]K M
K M 1 2
[E ]
1 0 [ S ]0
[E]0 [ES]
k1
[ S]0
[ S]0
KI
[I]0
[E]0 [ S]0 [ES] [ S]0 K M 1
K I
0I
d [P ]
k 2 [ES]
dt
[I]
K M (эфф ) K M 1 0
KI
[ES]
[E]0 [ S]0
[ I]
[ S]0 K M 1 0
KI
k 2 [E]0 [ S]0
max [ S]0
[I] [ S]0 K M (эфф )
[ S]0 K M 1 0
KI
0 0I
[ S]
0 max 0
[ S]0 K M
177.
10
[I]0>0
[I]0=0
Координаты Лайнуивера-Берка
1
0
1
max
1
1
K M (эфф )
KM
1
[ S ]0
0
KI
1
max
KM
max
1
[S]0
[I]0
K M (эфф ) / K M 1
Неконкурентное ингибирование
1. E + S
k1
k–1
ES
k2
P+E
2. E + I
3. ES + I
KI
[E][I]
[EI]
K
EI
ESI
[ES][I]
[ESI]
Если KI = K’ – ингибитор не конкурирует с субстратом, но уменьшает
скорость реакции из-за неактивности образующихся
на поверхности комплексов.
178.
0Imax( эфф ) [ S]0
max( эфф )
[ S]0 K M
1
1
KM
1
0I max( эфф ) max( эфф ) [S]0
max
[ I]
1 0
KI
1
0
[I]0
max
max( эфф )
[I]0=0
1
max( эфф )
max
1
KM
1
[ S ]0
0
Бесконкурентное ингибирование
E+S
k1
k–1
ES
k2
1
Координаты Лайнуивера-Берка
[I]0>0
1
KI
P+E
ES + I
KI
k3
k–3
[ES][I]
[ESI]
ESI
179.
[I]0>01
0
Координаты Лайнуивера-Берка
[I]0=0
1
max( эфф )
1
max
0I
1
K M (эфф )
1
KM
max( эфф ) [ S]0
[ S]0 K M ( эфф )
K M (эфф ) 1
1
1
0 I max( эфф ) max( эфф ) [ S ]0
1
[ S ]0
0
K M (эфф )
KM
[ I]
1 0
KI
180.
Автокаталитические реакцииA→B+C
A + (B) → B + (B) + C
B – катализатор
a – начальная концентрация A к моменту t0
(a–x) – концентрация A к моменту t
x – концентрация B к моменту t = изменение А
dx
k1 (a x )( x )
dt
dx
k1 (a x )(b x )
dt
k1dt
Добавим
B=b
(b<<a)
dx
1 dx
dx
(a x )(b x ) a b a x b x
a–x
a (b x )
1
ln
k1 t
a b b( a x )
1
2
3
b1 > b2 > b3
t
181.
a (b x )ln
(a b) k1 t Ut
b( a x )
e Ut
(a b) k1 U
a (b x )
e Ut b (a x ) a(b x )
b(a x )
e Ut b a e Ut b x a b a x
e Ut b a a b a x e Ut b x
x (a eUt b) b a (eUt 1)
b a (e Ut 1)
x
e Ut 1
x
Ut
a b e
b 1 r e Ut
в точке перегиба: x max
a+ b
max= k1 ×
2
max
2
a b a
2
2
x/b
r
b
a
(b a )
k1 (a x )(b x )
a2
k1
(b a)
4
Ut
182.
Время достижения максимальной скорости t½a b
a
b
1
1
2
t
=
-lnr×
lnr×
(b< < a)
(a b ) k1 t 1 / 2 1/ 2
ln
(a+ b) k1
a k1
b a a b
2
a b
a
x max
a (b x )
1
ln
k1t
a b b( a x )
2
( b a )
2
a
b+ x
ln
= ln + (a+ b)k1t
ax
b
b a
ln
x
a x
a
x
ln
ln
a k1 t
b
a x
ln
tg α = a k1
a x b
b x x
a b a
a
b
t
183.
lnx
b
a
ln a k1 t | x
a x
a
2
0 = ln
В точке перегиба
a/ 2
b
= ln + a k1 t 1/ 2
a- a/ 2
a
b
-ln = a k1 t 1/ 2
a
a
ln
a k1 t 1 / 2 a k1 t
a a
x
доля А за t
a
можно рассчитать
долю α
1
ln
a k1 t1/ 2 a k1 t a k1 t1/ 2 t
tИ – автокаталитическая реакция практически не идет
tИ – период индукции
1 И
1
t И t1/ 2
ln
a k1 И
И
xИ
a
И
1
t1/ 2
ln
a k1 1 И
xИ – кол-во прореагировавшего в-ва А ко времени tИ
184.
Правило Оствальда: сила каталитического действия кислотрямо пропорциональна их электропроводности
K a – константа диссоциации слабой кислоты
Аррениус:
1) Первичный солевой эффект:
Каталитическое действие кислоты увеличивается
при добавлении в раствор соли, не имеющей общего
иона с этой кислотой.
2) Вторичный солевой эффект:
Каталитического действие слабой кислоты также
увеличивается при добавлении в раствор ее соли.
Ионная сила раствора I 1 Ci Z i2
2
A+B
AB≠
k2
P (р–р)
A и B – ионы
k БT
k БT CAB
k æ
KC æ
h
h CA CB
aAB
CAB
AB
AB
K
KC
aA aB CA CB A B
A B
a
C
185.
k ækБT A B
K
k0 A B – уравнение Бренстеда–Бьеррума
h
AB
AB
k0 – константа скорости при бесконечном разбавлении (γ=1)
Дебай–Хюккель
A ZA
k k0 A B
AB
lg z A zB h I → средний коэф-т активности
B ZB
AB Z A ZB
k
A B
k0
AB
A B
k
lg lg
k0
AB
lg z A zB h I
h Z A2 Z B2 Z A Z B I 2h Z A Z B I
k
lg 1.02 Z A Z B I
k0
k
lg
k0
2
1.826 106
h
( T ) 3 / 2
Z A ZB 0
Z A ZB 0
I
Z A ZB 0
при 250С → h = 0.51 (H2O)
186.
D ED
i
Kинетические аспекты катализа
a
ΣDi – сумма энергий разрываемых связей
i
степень компенсации
kБ T
k æ
KC
F RT ln K
h
Δ F¹ 0(1-x)
Δ S¹
Δ U¹ 0(1-x)
kБ× T
kБ× T
k= æ
exp = æ
exp × c
× exp × c
h
RT
h
R
RT
æ – трансмиссионный коэф-т
kБT/h – эффективная скорость прохождения через барьер
x – число молекул в активном комплексе
x 1
RT
для газовой фазы: c 0(1 x ) 0
P
F 0(1 x )
S
U 0(1 x )
kБ T
kБ T
c
exp
c
k æ
exp
æ
exp
h
h
RT
R
RT
Ea
k
A
exp
S 0(1 x )
E
kБ T
a
RT
k æ
e exp
exp
c
h
R
RT
E U RT
S
A A0 exp
R
lg A lg A0 S
a
S const a E a
187.
Гетерогенный катализВещество С +
К (разные фазы)
– активные центры (АЦ)
Катализатор
АЦ – от 4% до 100% всей поверхности К
В кн.: Гетерогенный катализ (Боресков Г.К. –М. Наука 1986)
A – каталитическая активность К.
поверхность
A кат 0 (1 )
φ – доля объема системы К, недоступная для
0 (1 ) 0
реагирующих веществ.
A
к ат
a – удельная активность К.
A
a=
к ат
S – поверхность
S
S
188.
В промышленности:1.Переходные металлы. 4 группы:
А: Ti, V, Cr, Mn, Zn, Nb, Mo, Hf, Ta, W
B: Fe, Co, Ni, Tc, Ru, Re
C: Cu, Rb, Pd, Os, Ir, Pt
D: Ag, Au
Носители – основа
2. Катализаторы кислотно-основного типа
Кислые катализаторы
а. Нелетучие минеральные кислоты (H3PO4, H3BO3, SiO2, уголь, кизельгур)
б. Натуральные минералы (каолинит, бентонит)
в. Смешанные оксиды (Al2O3∙SiO2, Al2O3 ∙B2O3)
г. Соли сильных кислот (AlCl3, CuSO4, NiSO4)
Основные катализаторы
д. Неорганические соли и оксиды основного характера
(BaO, CaO, MgO, K2CO3, CaCO3)
Цеолиты: Me2/nO ∙Al2O3 ∙xSiO2 ∙yH2O
Промоторы (добавки): 1 – структурообразующие (препятствуют
увеличению размера кристалла)
2 – модифицирующие (влияют на структуру и
состав катализатора)
Отравление катализатора (у катализатора свой набор ядов)
Aотр.К A 1 C;
α – коэф-т отравления, С – концентрация яда
189.
адсорбатА
Изотермы адсорбции газов. Закон Генри.
А
Адсорбционное равновесие
Сa – концентрация адсорбата,
число молей в объеме адсорбционного слоя
адсорбент
γa – коэффициент активности адсорбата
в объеме адсорбционного слоя
С – концентрация адсорбата в газе
γ – коэффициент активности адсорбата в газе
Ca a
– коэффициент распределения или константа равновесия
C
(не зависит от концентрация адсорбата)
K f (T )
K
K C
Ca
a
Ca K C
C
Ca K
a
Ca min
a 1
Для идеальных газов: C(
a Va Ca S Ca
a
Ca
S
1
при невысоких давлениях
1
P
)=
V
RT
Ca
K
P
RT
а – кол-во молей адсорбата на 1 г. адсорбента;
S – поверхность адс. слоя; Va – объем адс. слоя;
τ – толщина адс. слоя; α – поверхностная концентрация
190.
a Va K C VaT = const
K
P – закон Генри
RT
Va const
K a ,P Va
a K a ,P P
K C
K
P K ,P P
RT
закон Генри
K
RT
K a ,P
K
RT
Са и α – определяются природой адсорбата и адсорбента.
C
a
– степень заполнения
a
Cam am m
Саm, аm и αm – соответствуют max заполнению поверхности
K
K
K
P a ,P P ,P P ;
Cam RT
am
m
А1
А2
K a,P
А1
α
am
K a,P
am
А2
K α,P
m
K α,P
α
P
m
P
191.
Теория адсорбции Ленгмюра (хемосорбция)Основные предпосылки
1. Одинаковые адсорбционные центры
2. Независимость энергии хемосорбции от степени заполнения
3. Слой – мономолекулярный
Механизмы сорбции
I. A(г) + S
kадс
kдес
(A…S)адс
S – свободный адсорбционный центр;
(A…S)адс – заполненный адс. центр;
kадс, kдес – константы скорости адсорбции
и десорбции
a – величина адсорбции при Р;
amax – предельная величина адсорбции
a
– степень заполнения
am
адс k адс P (1 )
(1 – θ) – доля свободной поверхности
дес kдес
процесса адс дес k адс P (1 ) kдес ;
k адс P (1 ) kдес
kадс P kадс P kдес
K
процесса 0 (при равновесии)
k адс
– константа равновесия адсорбции
k дес
192.
K P K PK P
1 K P
a
am
Изотерма Ленгмюра
K b e
Gадс
RT
e
Sадс
R
e
Hадс
RT
b0 e
Q
RT
b – адсорбционный коэффициент вещества;
b0 – адсорбционный коэффициент вещества, связан с энтропией;
ΔH - энтальпия адсорбции; Q – теплота адсорбции.
Термодинамический вывод уравнения Ленгмюра
A(г) + S
A S AS
(A…S)адс
A A RT ln
PA
P
S S RT ln(1 )
AS AS RT ln
am a am
a am
- Свободные адсорбционные
центры
- Занятые адсорбционные центры
A
При равновесии: A RT ln P S RT ln(1 ) AS RT ln
RT ln P A RT ln(1 ) RT ln AS A S
P A (1 )
RT ln
Gадс
G RT ln K
a
K P
Изотерма Ленгмюра
K
1 K P am
P (1 )
193.
1. K∙P<<12. K∙P >>1
K P
1 K P
K P
1 K P
K P (з-н Генри для газов)
1
1.0
P
K P
1 K P
1
2
1
K
K P
P
0
am K P
1 K P
1 1 K P
1
1
a am K P am K am P
II. A2 + –S–S–
адс
kадс
kдес
k адс P (1 A )2
При равновесии:
A A
–S–S–
1 1
~
a p
Диссоциация A2 на атомы
дес kдес 2A
адс дес
1
2
a
K P
am 1 K P
a
1
K
K P 1
k адс P (1 A )2 kдес 2А
194.
K P 121
1 K P 2
1
K P 2
1
1. K∙P<<1
K P
1
2
K
k адс
k дес
2. K∙P >>1
1
III. A и B – адсорбаты (конкуренция)
θA и θB – степени заполнения A и B (конкуренция)
PA и PB – парциальные давления A и B
адс ,A k адс ,A PA (1 A B );
дес ,A kдес ,A A
дес ,B kдес ,B B
адс ,B k адс ,B PB (1 A B )
адс дес
адс ,B
K A адс ,A ; K B
дес ,B
дес ,A
1. KA∙PA и KB∙PB <<1
θA = PA × K A ; θB = PB × KB
2. KA∙PA >> 1 + KB∙PB
A 1 и B
PA
PB
195.
Полимолекулярная адсобцияТеория БЭТ | Брунауэр, Эммет, Теллер
1) газ + свободная поверхность
2) газ + единичные комплексы
3) газ + двойные комплексы
единичные комплексы
двойные комплексы
тройные комплексы
, , ...– доли поверхности (единичные, двойные, тройные комплексы)
a am 2 3 ...
K
; K
;
P 0
P
K
P
K
P (1 )
θ0 – доля свободной поверхности
K K , K , K
Предполагают: K K ...KL
K P K L P
2
P
2
K P K L P
PS
P
PS
KP
= θ
1+ K P
Изотерма
Ленгмюра
KL – константа
конденсации
K L 1/ Ps
насыщенный пар (PS)
жидкость
196.
3P
3
K P K L P
PS
2
P
P
a am K P 0 1 2
3 ...
PS
PS
a am 2 3 ...
2
P
P
0 ... 0 1 K P 1
... 1
PS PS
суммарное заполнение 1 слоя
прогрессии:
Σ убывающей геометрической
P / PS 1
2
P
Производная по
PS
P P
1
1
...
;
PS PS
1 P / PS
a am
K P 0
1 P / PS 2
2
P
P
1
1 2
3 ...
2
PS
P
1
P
/
P
S
S
0
1 P / PS
1
K P
1 K P P / PS
1
1 P / PS
197.
Pam K P
a
1 P / PS 1 K P P / PS
am C P / PS
1 P / PS 1 (C 1) P / PS
m C P / PS
1 P / PS 1 (C 1) P / PS
1 P
K L PS
K
C
KL
a
C P / PS
1 P / PS 1 (C 1) P / PS
Уравнения БЭТ
P / PS
a (1 P / PS )
1. P / PS min и C 1
a
K P
Ленгмюр
am
1 K P
C 1
am C
1
b
am C
tg
2. Линейная форма:
P / PS
1
C 1 P
a (1 P / PS ) am C am C PS
PS P
PS
b
P / PS
198.
Кинетика гетерогенно-каталитических реакцийгаз или жидкость
1. Диффузия
2. Адсорбция
3. Химическая реакция на катализаторе
4. Десорбция продуктов
5. Диффузия
A(г) + S
I. Мономолекулярная реакция
k
P(г) + S
(A…S)адс
S – общая площадь поверхности
dn
S dt
dn
k A k
S dt
= k θA =
газ или жидкость
n – число молей A
k K PA
1+ K PA
k S k
θA =
K • PA
1+ K • PA
S const
199.
при K PA 10 порядок по А
k K PA при K PA 1
1 порядок по А
kк аж= k K
o
E каж E а .к . H адс
Еист.гет
Е
Еадс
= k θA =
Екаж.гет
= k
о
H адс
H ро
Едес
о
Hдес
коорд.
Продукты уменьшают поверхность катализатора
A(г) + S
(A…S)адс
k
(В…S)адс
B(г) + S
лимитирующая стадия
A
K A PA
1 K A PA K B PB
k A
k K A PA
1 K A PA K B PB
k K PA
1+ K PA
200.
В – играет роль ингибитораЕсли
k
KA PA 1 KB PB
0 порядок
= k θ =
A
k K P
A
1+ K P + K P
A
Если
K A PA KB PB 1
Если
KA PA 1 KB PB (KB PB 1)
k KA PA
k K A PA
P
k каж A
K B PB
PB
II. Бимолекулярные реакция
А+В
P1 + P2
Механизм Лэнгмюра-Хиншелвуда
A(г) + S
(A…S)адс
(A…S)адс + (В…S)адс
(P…S) адс
В(г) + S
(В…S)адс
(P…S) адс
P1 + P2 + S
A и B – конкурируют за адсорбционные центры
A
K A PA
1 K A PA K B PB
B
A
1 порядок
E каж Е a ,k H адс ( A) H адс (B)
K B PB
1 K A PA K B PB
A
B
B
201.
В – играет роль ингибитораЕсли
k
KA PA 1 KB PB
0 порядок
= k θ =
A
k K P
A
1+ K P + K P
A
Если
K A PA KB PB 1
Если
KA PA 1 KB PB (KB PB 1)
k KA PA
k K A PA
P
k каж A
K B PB
PB
II. Бимолекулярные реакция
А+В
P1 + P2
Механизм Лэнгмюра-Хиншелвуда
A(г) + S
(A…S)адс
(A…S)адс + (В…S)адс
(P…S) адс
В(г) + S
(В…S)адс
(P…S) адс
P1 + P2 + S
A и B – конкурируют за адсорбционные центры
A
K A PA
1 K A PA K B PB
B
A
1 порядок
E каж Е a ,k H адс ( A) H адс (B)
K B PB
1 K A PA K B PB
A
B
B
202.
k A Bk K A K B PA PB
1 K A PA K B PB 2
1. A и B – слабо адсорбируются (K A PA 1, KB PB 1)
k KA KB PA PB
2. A адсорбируется сильно, B – слабо ( K A PA 1 KB PB )
k K B PB
K A PA
3. B адсорбируется сильно, A – слабо ( KB PB 1 K A PA )
k K A PA
K B PB
Механизм Или-Ридиела
A(г) + S
(A…S)адс
(A…S)адс + В
k A PB
P+S
B – в газовой фазе
k K A PA PB
1 K A PA K B PB