


")

")



")

")



















 Медицина
МедицинаПохожие презентации:






")



Патофизиология системы Эритрона
1.
ПАТОФИЗИОЛОГИЯ СИСТЕМЫЭРИТРОНА
КАФЕДРА ПАТОЛОГИИ
2.
• АнемииАнемия – клинико-гематологический синдром,
характеризующийся ↓ кол-ва эритроцитов
(N мужчины 5,5±0,9·1012/л; женщины 4,8±0,6·1012/л) и/или
концентрации гемоглобмна (N мужчины 160±20г/л;
женщины 140±20г/л) в ед. объема крови.
• Главная патофизиологическая суть анемии
↓ кислородной емкости крови, приводящей к гипоксии
гемического типа, а затем, и к тканевой.
• С тканевой гипоксией связаны дальнейшие нарушения
жизнедеятельности органов и тканей, в особенности
высокоаэробочувствительных.
3.
4.
5.
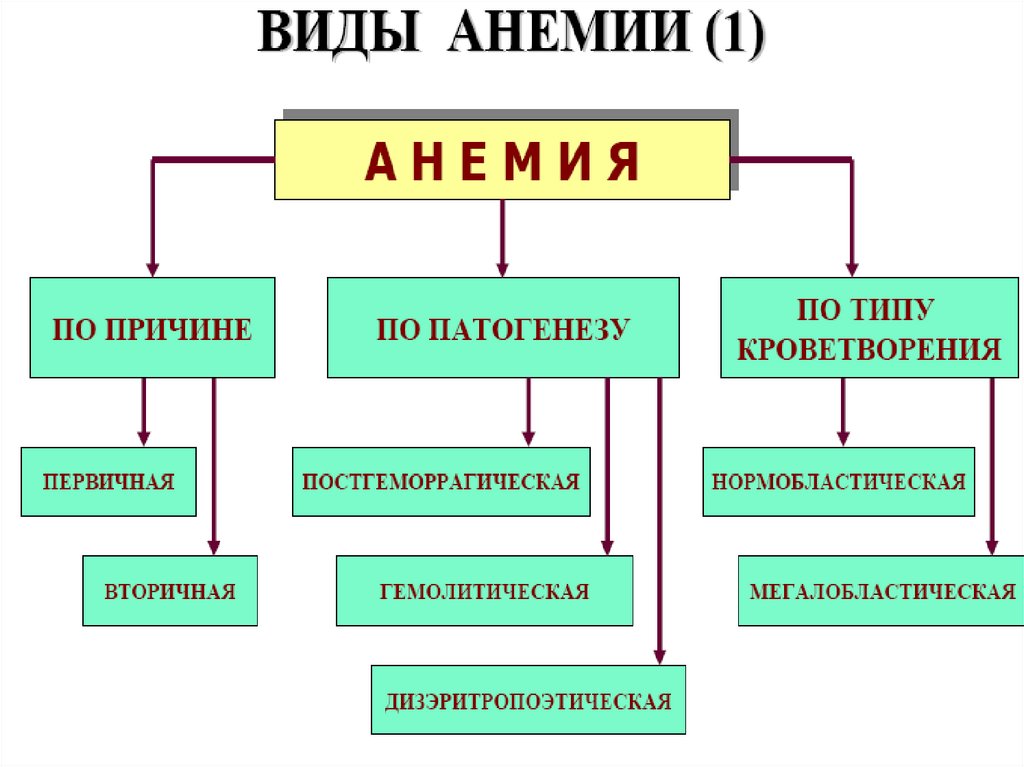
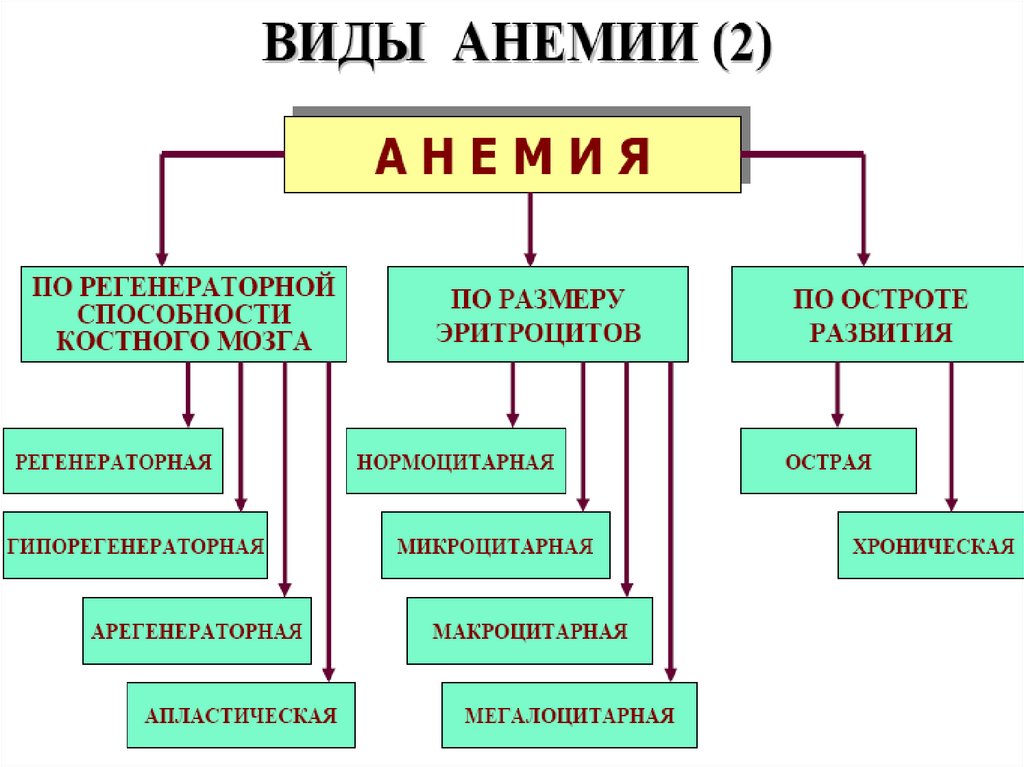
ВИДЫ АНЕМИИ (3)По цветовому показателю
• Нормохромные (0,86-1,0)
• Гиперхромные (>1,0)
• Гипохромные (<0,86)
По конц-ции Нв степень анемии:
• 80-100 г/л легкая
• 60-80 г/л средняя
• 60 г/л тяжелая
6. Острая постгеморрагическая анемия (ОПГА)
• Потеря 15-25% ОЦК формирует легкую ОПГА• Потеря 25-35% ОЦК формирует ОПГА средней
степени тяжести
• Потеря 35-50% ОЦК формирует тяжелую
ОПГА
• Потеря более 50% ОЦК летальна
• Ведущие патогенетические механизмы ОПГА
• ↓ ОЦК → гематоциркуляторные расстройства →
гипоксия → нарушения всех видов аэробного
метаболизма
7. Фазы течения острой ПГА
• Рефлекторная фаза компенсации. Картина крови не отличается отнормы (нормоцитемическая гиповолемия). Ht, Э, Нb - норма.
«Скрытая анемия» - нормохромная, нормоцитарная. Клинические
проявления характерны для коллапса: ↓ АД, бледность,
тахикардия, тахипноэ. Возникает сразу после кровопотери.
• Гидремическая фаза компенсации. Характеризуется направленностью
на восстановление ОЦК и АД. (полидипсия и питье воды,
гемодилюция – перемещение тканевой жидкости в сосуды, ↓
диуреза, активация РААС, усиление реабсорбции Na и воды).
После восстановления ОЦК (2 - 4 сутки) анемия носит
нормохромный характер. Гипоксия стимулирует выделение
эритропоэтинов почками ►↑ эритропоэз.
• Фаза костно-мозговой компенсации. В крови ретикулоцитов до 30-40%
(гиперрегенераторная анемия). ЦП <0,85 (гипохромная анемия),
скорость синтеза Нb отстает от темпа пролиферации клеток. В
костном мозге – явления интенсификации эритропоэза — ↑
количество эритробластов и ретикулоцитов. Длится до 14 дней
при тяжелой форме.
8. Виды гемолитических анемий (ГА)
9. Этиология приобретенных ГА
1. Аутоиммунный гемолиз эритроцитов «тепловыми» аутоантителами при изменении комплекса МНС лекарственными препаратами (a-метилдопа, пенициллины, цефалоспорины, сульфаниламиды и др.) и в ходе системной патологии(системная красная волчанка, хронический лимфолейкоз, лимфома и др.).
«Тепловой» гемолиз реализуется макрофагами селезенки. Аутоиммунный гемо
лиз «холодовыми» аутоантителами индуцируется инфекцией (Mycoplasma
pneymoniae, вирус Эпштейна-Барр) и осуществляется преимущественно в сосудистом русле.
2. Изоиммунный гемолиз (гемотрансфузионный) эритроцитов развивается при
резус-конфликтной ситуации (гемолитическая болезнь новорожденных) и несовместимости по АВО-системе. Реализуется макрофагами селезенки.
3. Механический гемолиз эритроцитов реализуется искусственными клапанами
сердца, протезами сосудов и т.д. в кровеносном русле.
4. Токсический гемолиз эритроцитов реализуется гемолитическими ядами грибов (афалотоксин бледной паганки), мышьяком, свинецом, бактериальными
токсинами (клостридиальный, стафилококковый, стрептококковый), токсинами простейших (плазмодий малярии). При острых токсических ГА разрушение эритроцитов происходит в сосудах, а при хронических - преимущественно
в селезёнке.
10. Патогенез приобретенных ГА
Приобретенные ГА характеризуются гемической гипоксией и интоксикацией,обусловленной метаболитами гемоглобина. ↑свободного билирубина вызывает би
лирубиновую энцефалопатию и слабовыраженную желтуху. Ферритинемия нередко вызывает сосудистый коллапс. Усиленный фагоцитоз гемоглобина макрофагами селезенки и последующий его метаболизм до гемосидерина, вызывают
гемосидероз органа, который всегда увеличен и болезнен (спленомегалия). В
моче появляется избыток пигментов билирубинового обмена и она приобретает
цвет «тёмного пива». Их повышение в желчи и кале носит название плейохромия. Канальцы почек забиваются гемоглобиновыми цилиндрами (гемоглобинурийный нефроз). Агглютинаты эритроцитов, микротромбы в сосудах могут нарушать кровообращение во внутренних органах.
Общим гематологическим признаком приобретенных ГА является ретикулоцитоз, что отражает компенсаторное регенераторное напряжение (состояние)
эритропоэза. В красном костном мозге этапы созревания эритроцитов не нарушены, однако цветовой показатель снижен (гипохромная анемия).
В периоды обострений (гемолитические кризы), или при первично тяжелом
течении гематологическая картина характеризуется явлениями гипорегенерации
в виде анизоцитоза и микроцитоза. В красном костном мозге развивается гипоплазия.
11. Костный мозг при гипопластической анемии
«Чисто белоклеточный» костный мозг с частично сохраненнымлейко - и тромбоцитопоэзом.
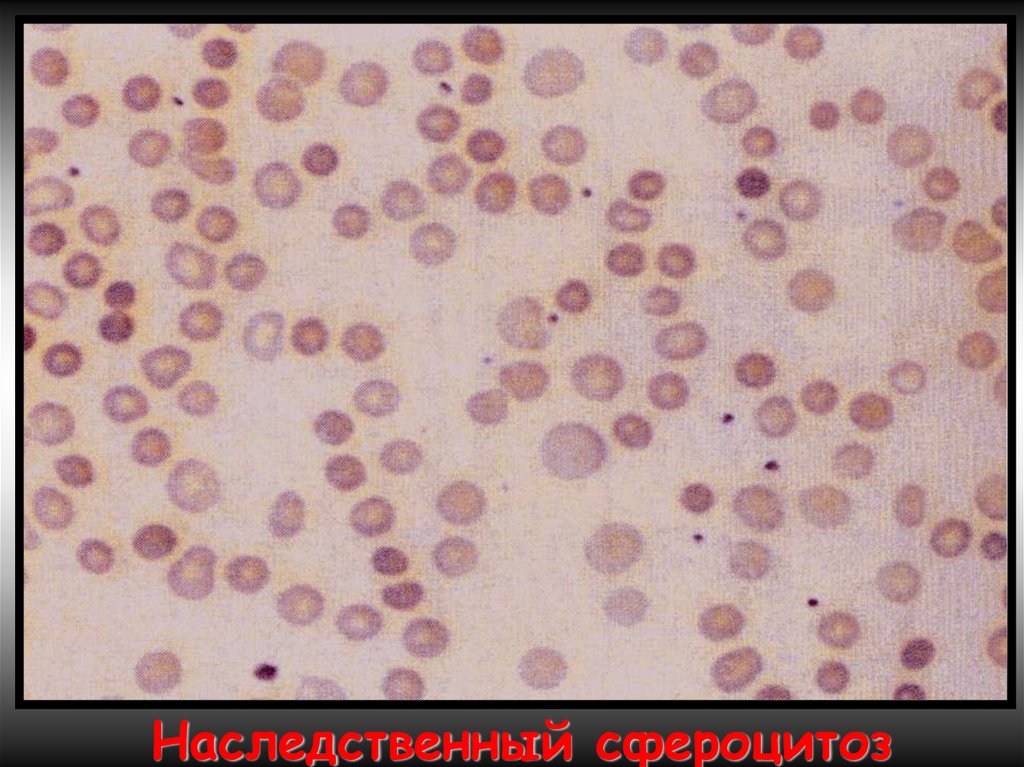
12. Мембранопатии (эритроцитопатии)
Патогенез – врожденные дефекты клеточной мембраны. Нар-ие синтеза белков(спектрин, анкерин и др.)
Семейный наследственный микросфероцитоз
(болезнь Минковского-Шоффара).
Наследуется аутосомно-доминантно. Мембранопатия обусловлена
значительным ↓ содержания белка спектрина, нарушением связывания
его с другими белками мембран. Структура самого спектрина при
микросфероцитозе изменена. Все это обусловливает ↑ проницаемости
мембраны эритроцитов для ионов Nа+,Са²+ и накопление их избытка, а
также жидкости в гиалоплазме. Гипергидратированные эритроциты
приобретают сферическую форму. Это ↓ пластичность мембран
эритроцитов, их способность к деформации в микрососудах. Проходя
через селезеночные синусы, они не могут деформироваться, теряют
часть поверхности и превращаются в сфероциты малого размера,
резко ↓ их продолжительность жизни (до 6-8 суток).
Характеризуется длительным латентным течением часто с одним
симптомом желтухи с микросфероцитозом. Провоцируют обострение
переохлаждение, переутомление, инфекции. В остром периоде спленомегалия и желтуха с уробилирубинемией и уробилинурией, а
также ↑t°. Часто возникают трофические язвы (следствие
микротромбов при гемолизе).
13.
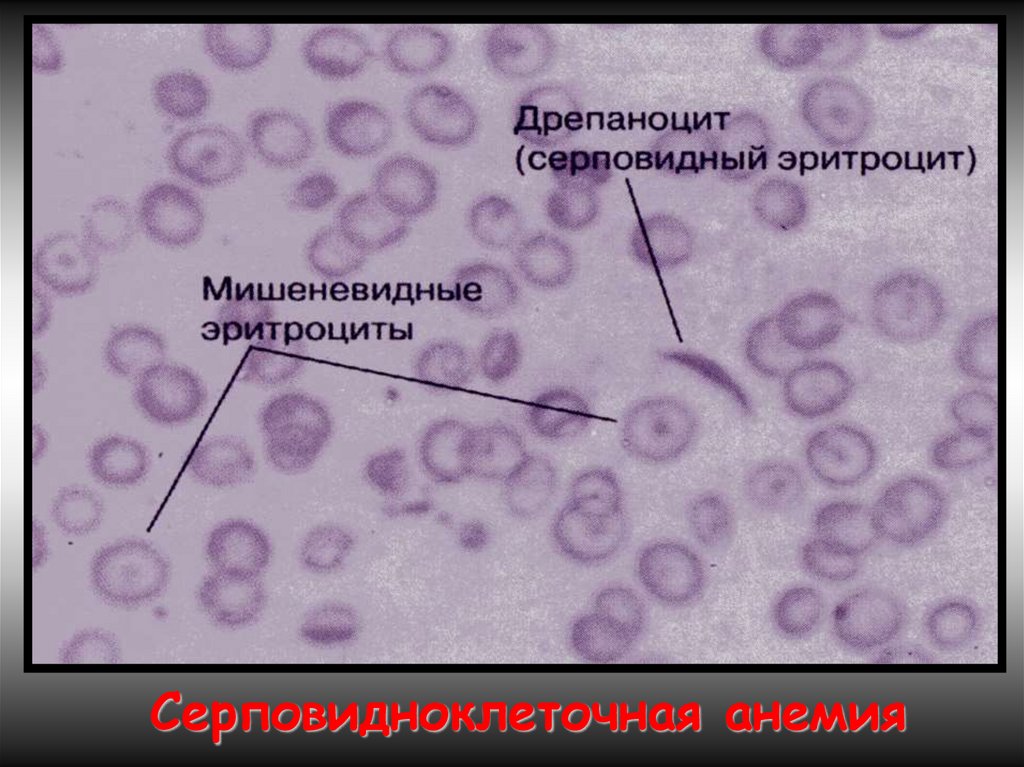
Наследственный сфероцитоз14. Гемоглобинопатии
Серповидно-клеточная анемия.• Патогенез — генетический дефект в структурном гене ⇒
нарушается структура β-цепей глобина в связи с заменой
одной аминокислоты (чаще глютамина) на другую (валин).
• В основе образования серповидных клеток лежит
свойство HbS полимеризоваться при переходе в
восстановленную форму. Образующиеся полимеры
(длинные нити) группируются в тактоиды, они
изменяют форму и вид эритроцитов. Полимеризация
восстановленного HbS связана с его низкой
растворимостью (в 100 раз ↓ растворимости HbA).
Образование тактоидов зависит от концентрации HbS в
эритроците и парциального давления кислорода в
крови.
• ↓Нв до 30-50 г/л, ретикулоцитоз, ↑ сод-ия железа в
сыворотке, гипохромия.
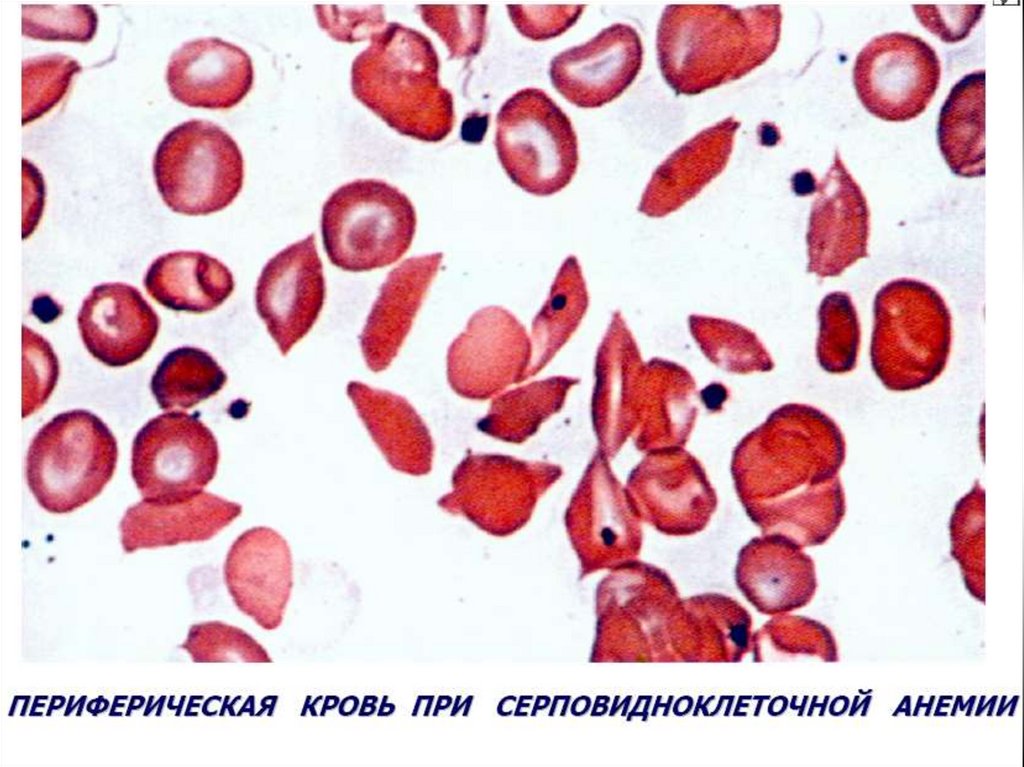
15.
Серповидноклеточная анемия16.
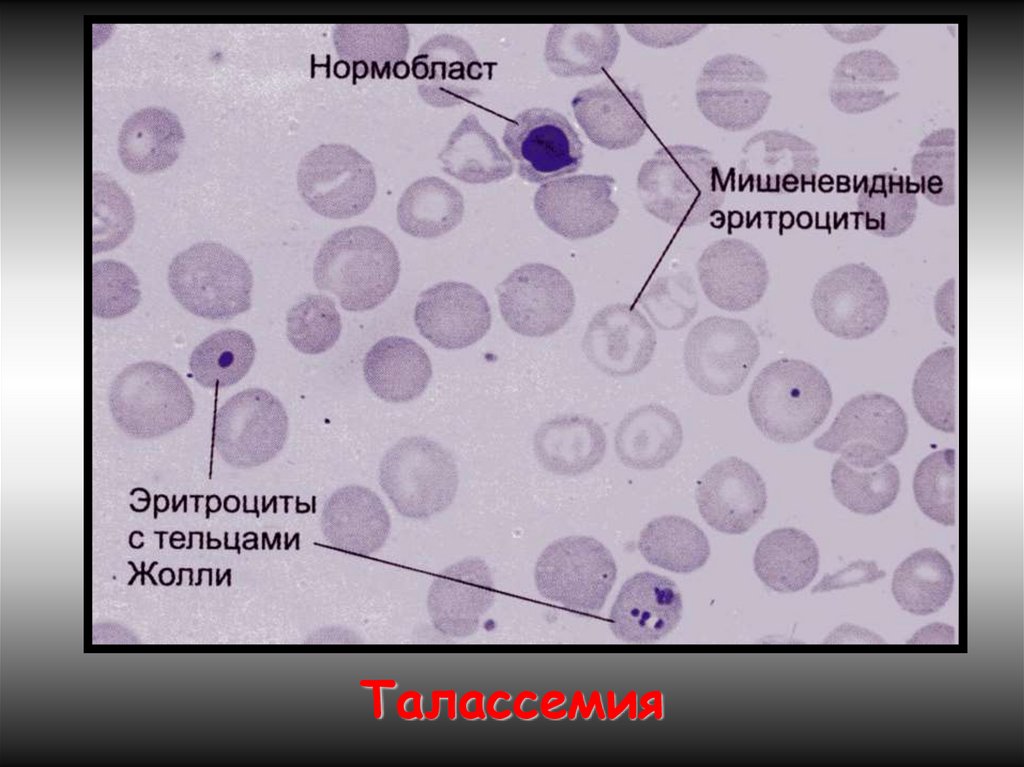
17. Талассемии (б. Кули-Кули)
• Талассемия (греч. talasia — море) ⇒ дефицит HвA безкачественных нарушений его глобиновых цепей.
• Патология генов-регуляторов ⇒ в эмбриогенезе не
происходит норм. переключения синтеза глобиновых
цепей, обр-ся аномальный Нв в ущерб синтезу HbA.
• Две группы: α-талассемия и β-талассемия.
• При α-талассемии - частичная или полная делеция αглобиновых генов-регуляторов → ↓ синтез этих цепей.
Их недостаток комп-ся в эмбр. периоде избыточным
синтезом ץ-глоб. цепей (Нb Bart's). После рождения
недостаток α-глобиновых цепей восполняется
синтезом β-глобиновых цепей (HbH). (HbBart's и HbH маркеры α-талассемии).
• Клиническое проявление α-талассемии — гипоксия из-за
высокого сродства HbBart's и HbH к О2. Анемия
усугубляется ↑ разрушением эритроцитов в селезенке.
18.
β-талассемия.Β0 β+ талассемии.
Патогенез:
2 механизма: -Нв синтезируется меньше,
гипохромия.
-↓жизни эритроцитов из-за
несбалансированного синтеза цепей
Нв.
Итог: неэффективность эритропоэза.
19.
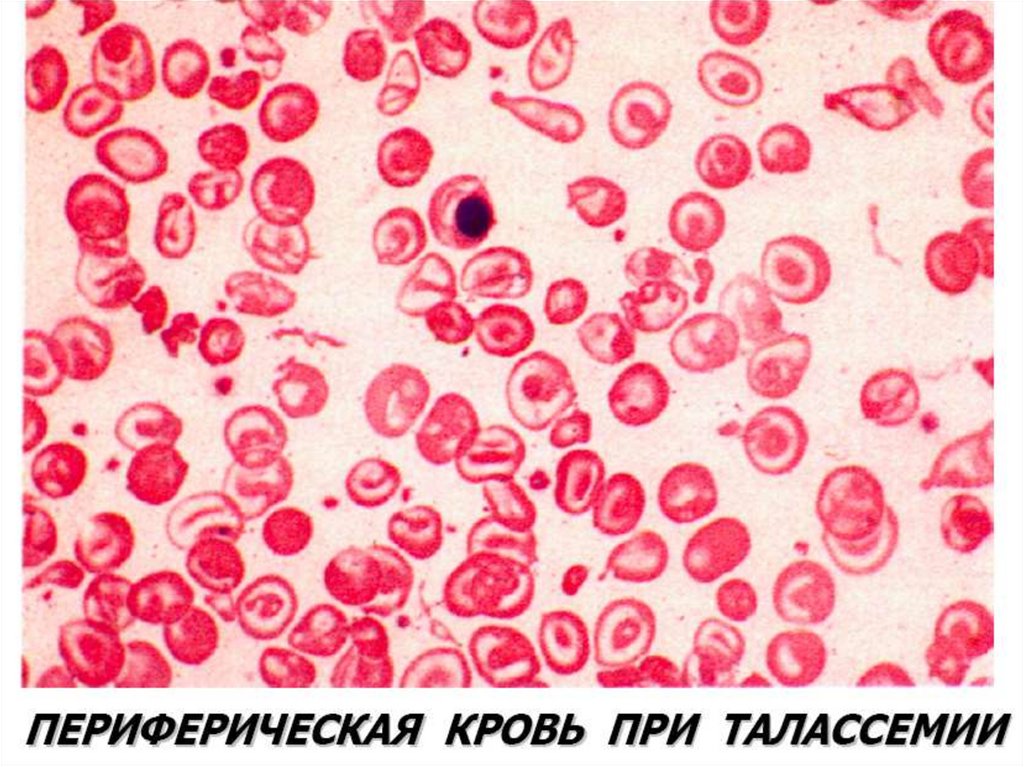
Талассемия20.
21. Энзимопатии
Наиболее часто обнаруживают следующие дефекты:
дефекты активности ферментов гликолиза: пируваткиназы, гексокиназы,
фосфофруктокиназы и др. В эритроцитах ведущим путем ресинтеза АТФ
является гликолиз. →→ недостаток энергии АТФ обусловливает
нарушение трансмембранного переноса ионов. Развивается их дисбаланс
► гипергидратация и набухание эритроцитов;
↓ активности энзимов пентозофосфатного цикла. В ходе его реализации
образуется восстановленная форма НАДФ, использующаяся для
восстановления глютатиона. Восстановленный глютатион — компонент
антиоксидантной системы эритроцитов► при таких энзимопатиях имеет место
разрушение липопротеидных комплексов мембран;
дефицит ферментов системы глютатиона (глютатионсинтетазы,
глютатионредуктазы и т.д.).
Последствия активация липоперекисных реакций в эритроцитах,
нарушения целостности их мембраны.
Пример: гемолитическая анемия, связанная с недостаточностью в
эритроцитах Г-6-ФД. Гемолиз провоцируется приемом лекарственных
препаратов (сульфаниламиды, антипиретики, анальгетики и др). При ↓ Г-6-ФД
эритроциты быстро утрачивают минимум имеющегося восстановленного
глутатиона и быстро стареют
Анемия: нормохромная, нормоцитарная, регенераторная.
22.
23. ДИЗЭРИТРОПОЭТИЧЕСКИЕ АНЕМИИ
1. Обусловленные нарушением эритропоэза в связи спреимущественным повреждением стволовых клеток:
гипо- и апластические анемии.
2. Обусловленные нарушением эритропоэза в связи с
преимущественным повреждением клетокпредшественников миелопоэза и/ или
эритропоэтинчувствительных клеток:
• вследствие нарушения синтеза нуклеиновых кислот
эритроплазмоцитов (мегалобластические): витамин В12 —
(фолиево) дефицитные анемии; фолиеводефицитные
анемии.
• в результате нарушения синтеза гема:
железодефицитная анемия; порфириндефицитная;
• вследствие нарушения синтеза глобина: талассемии; при
нар-ии первичной структуры цепей глобина (серп.-клеточ.
ан.).
• При нар-ии регуляции деления и созревания
эритроид.клеток.
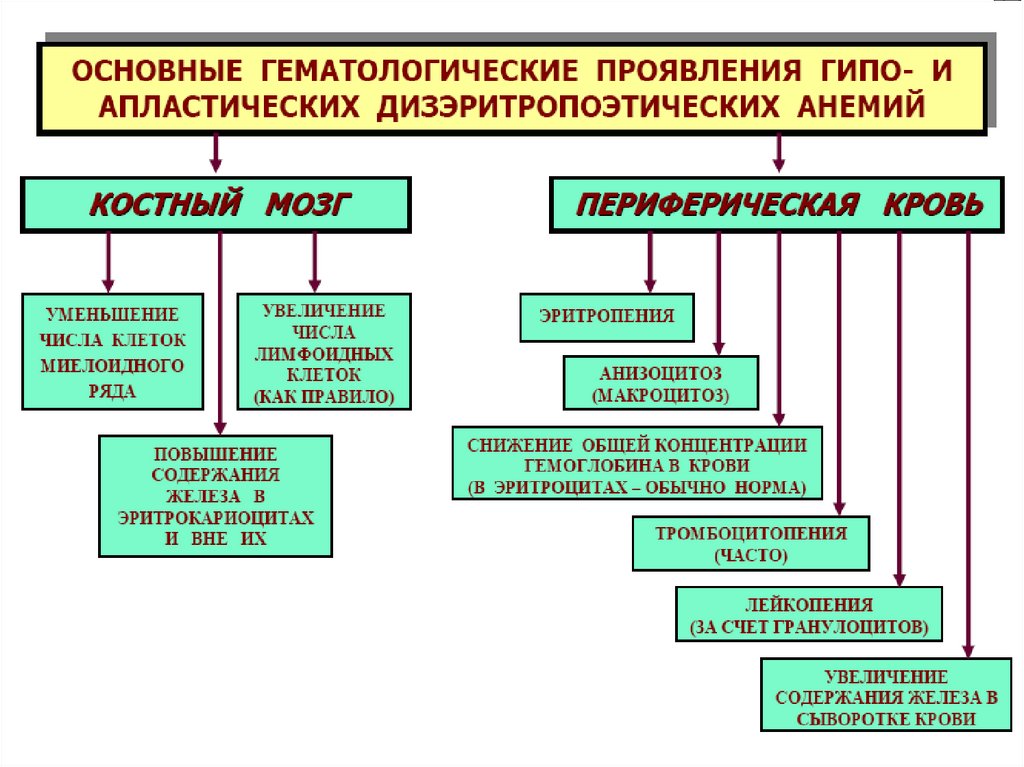
24. 1.Гипо- апластические анемии
Являются синдромами др.пат. процессов.По происхождению:
• Приобретенные (вторичные) под действием агентов физ.,
хим., биол.природы.
Патогенез – нар-ие пролиферации и/или гибель стволовых
гемопоэтич. клеток→анемия.
Проявления – гипоксия, кровотечения и кровоизлияния (из-за
↓сверт.крови), инфекции.
• Наследуемые (первичные).
Анемия Фанкони. (Аутосомно-рецессивно).
Патогенез – нар-ие процессов репарации ДНК стволовых
клеток, ↑мутабельности. В целом не ясен,
предположительно – иммунолог.супрессия, либо аномалии
стволовых клеток.
Проявления: гипоксия, тромбоцитопения, лейкопения
(инфекции).
25.
26. 2. Обусловленные нарушением эритропоэза в связи с преимущественным повреждением клеток- предшественников миелопоэза и/или
эритропоэтинчувствительных клеток.В12 —(фолиево) дефицитные анемии и фолиево-дефицитные анемии
(мегалобластические).
Дефицит В12 и фолиевой к-ты. Нар-ся синтез ДНК. В12 (кобаламин) и
фолаты – коэнзимы в процессе синтеза ДНК. Их недостаток →дефект
созревания ядра, нар-ие процессов деления. Асинхронность
созревания цитоплазмы и ядра. Аномальность кроветворения.
Для всасывания В12 необходим ф.Кастла.
2 формы В12:
• метилкобаламин (с его помощью фолиевая к-та превращается в
тетрагидрофолиевую; при его отсутствии не обр-ся предшеств.ДНК,
процесс обрывается, торможение наз-ся «капкан метилфолата»).
• аденозинкобаламин – дефицит ведет к накоплению метилмалоната
и пропионата → избыток аномальных ЖК в липидах нервных структур
→ неврологическая симптоматика.
27.
Клиническая триада при дефиците В12:• Мегалобластическая анемия;
• Глоссит Хантера;
• Фуникулярный миелоз.
Патогенез В12 — (фолиево) дефицитных анемий:
Анемия Аддисона-Бирмера.
Аутоиммунное заболевание. Цитотоксический тип поражения
слизистой желудка с потерей париетальных клеток его
фундального отдела. АТ 3 типов: I тип - блокирующие
(блокируют связывание В12 с ф.Кастла), II тип – нар-ие
связывания комплекса с рецептором, III тип – против
микроворсинок париетальных клеток.
Проявления мегалобласт.анемий:
Общие для В12 (фолиево) и анемии при дефиците фолатов:
связаны с нарушением синтеза ДНК (изменения в КМ,
крови, ЖКТ).
Различия: фолатная недостаточность не связана с потерей
ф-ии аденозин-кобаламина в синтезе ЖК и расстройствами
в периферич. и ЦНС.
28.
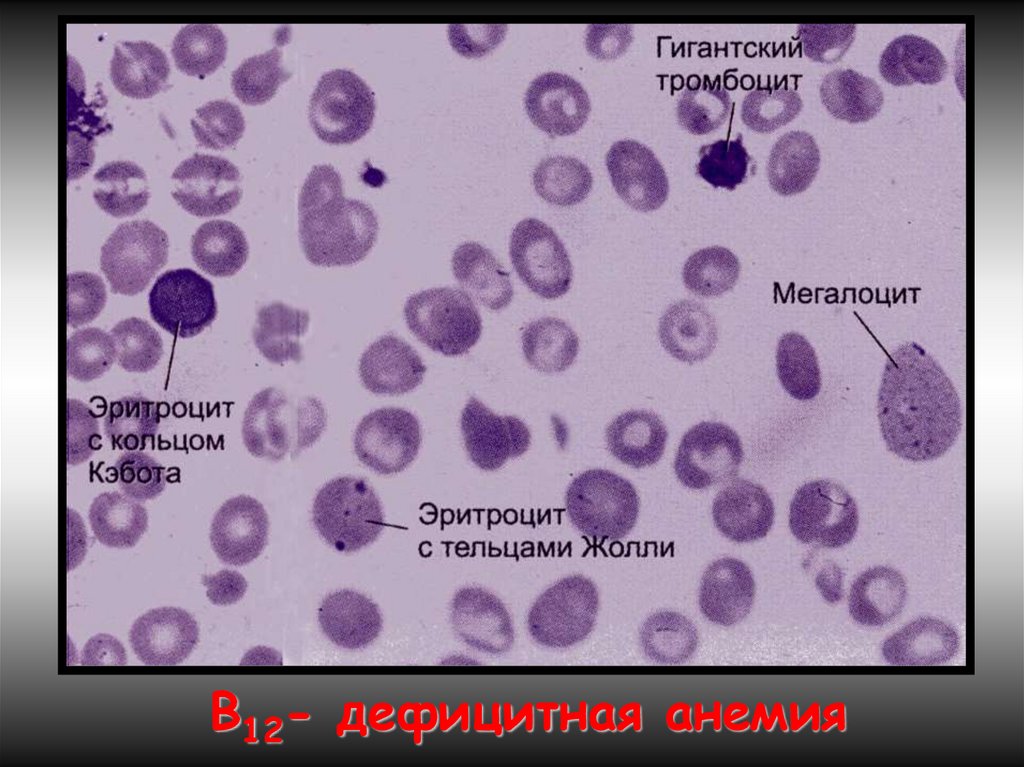
Проявления мегалобласт.анемий:В костном мозге обнаруживаются мегалобласты (d > 15 мкм)
«синий» КМ, а также мегалокариоциты. Нарушается
дифференцировка и других клеток миелоидного ряда.
В периферической крови значительно ↓ число эритроцитов,
иногда до 0,7-0,8 х10¹²/л. Они большего размера, овальной
формы, без центрального просветления. Встречаются
мегалобласты. В эритроцитах - остатки ядерного вещества
(тельца Жолли) и нуклеолеммы (кольца Кебота).
Характерны анизоцитоз и пойкилоцитоз. ЦП=1,1–1,3. Нb в
крови
существенно
↓
(из-за↓числа
эритроцитов).
Количество ретикулоцитов чаще ↓, как правило,
наблюдается лейкопения (за счет нейтрофилов). В связи с
↑гемолизом — билирубинемия.
Мегалобластические анемии хар-ся как гиперхромные, макромегалоцитарные, гипо- арегенераторные.
29.
В12- дефицитная анемия30.
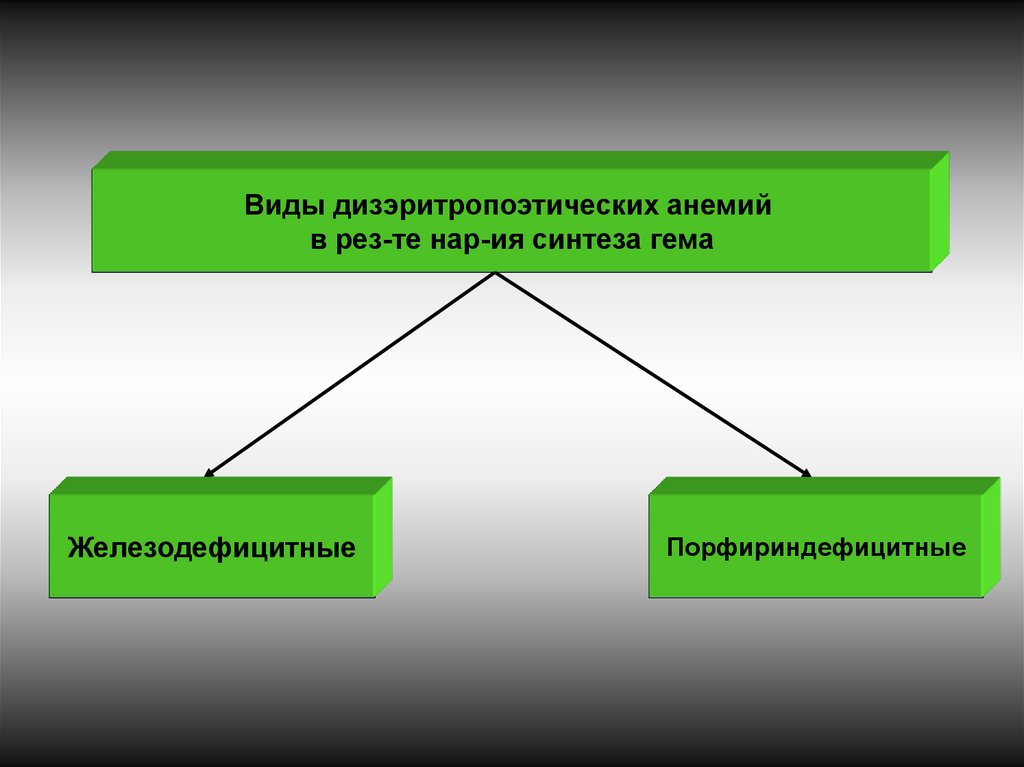
Виды дизэритропоэтических анемийв рез-те нар-ия синтеза гема
Железодефицитные
Порфириндефицитные
31.
32. Железодефицитные анемии
¾ всех анемий. Макс. – беременные и кормящие матери.Железо организма – функциональное (в Нв) и пул хранения
(гемосидерин и ферритин). Ферритин – комплекс железа с
белком (во многих тканях, много в макрофагах). Часть
ферритина в макрофагах деградирует с образованием
гемосидерина (гранулы).
Индикатор дефицита или избытка железа – сывороточный
ферритин.
Осн.причина ЖДА – хроническая кровопотеря.
Стадии дефицита железа:
1. Скрытый дефицит.
2. Латентный дефицит.
3. Постоянный дефицит.
4. Выраженный дефицит.
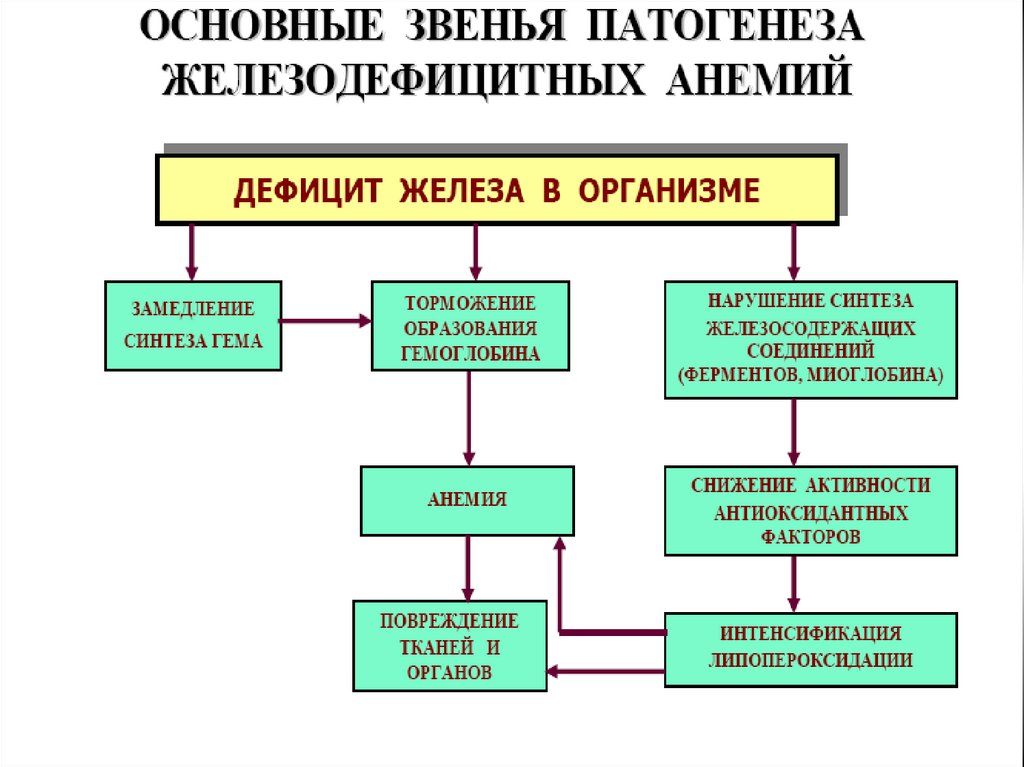
33. Патогенез ЖДА
По мере истощения лабильного депо железа в гепатоцитах, всеболее угнетается синтеза гема в гемоглобине. Одновременно
снижается активность большой группы железосодержащих
внутриклеточных ферментов: дегидрогеназ, цитохромов, каталаз и
пероксидаз, что ухудшает эффективность клеточного дыхания,
сопряженного с ним синтеза макроэргов и антиоксидантную защиту
клеток. В результате подавляется эритропоэз и снижается осмотическая резистентность эритроцитов, которые, повышенно разрушаясь,
усугубляют патологию.
В результате эритропоэз становится неэффективным, гипорегенераторным: в периферической крови уменьшается количество
нормоцитов, и появляются дегенеративные эритроциты: пойкило - и анизоциты с доминированием микроцитоза и гипохромии
(Ц.п. < 0,86).
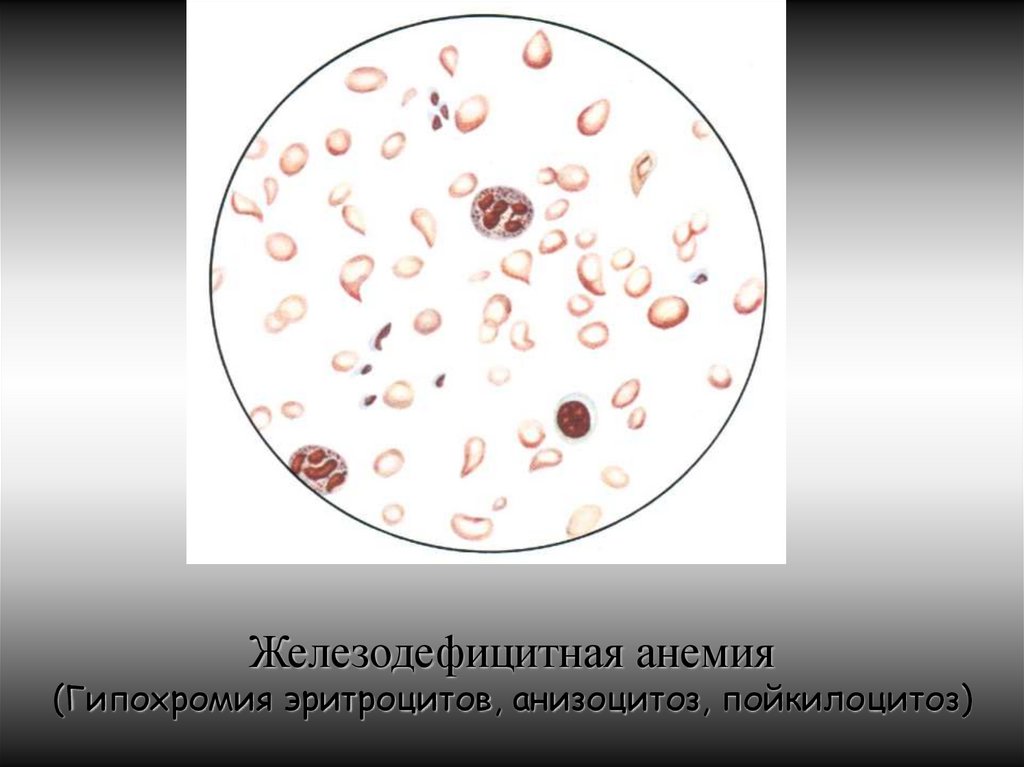
34.
Железодефицитная анемия(Гипохромия эритроцитов, анизоцитоз, пойкилоцитоз)
35. Патогенез ЖДА
При снижении содержания гемоглобина в крови до 6,8 - 6,6 ммоль/л, то ЖДАсчитают легкой степени тяжести, а менее 4,3 ммоль/л – тяжелой. Тяжелая ЖДА
проявляется дегенеративной панцитопенией и сопутствующими иммунодефици
тами (незавершенный фагоцитоз, ↓синтеза иммуноглобулинов, ↓активности лизоцима и β-лизина. На их фоне легко возникают и тяжело протекают инфекционные
заболевания.
ЖДА формирует гемическую, а затем и тканевую гипоксию, усугубляющих
неэффективность гемопоэза, и вызывающих метаболические и функциональные
расстройства в высокоаэробозависимых клетках: эпителии слизистых, кардиомиоцитах, нейронах.
Течение ЖДА – рецидивирующее: периоды обострений проявляются бледностью кожи, слабостью, физической утомляемостью, головокружениями, шумом в
ушах, обмороками. Дыхание частое, поверхностное. Обнаруживается тахикардия
и признаки левожелудочковой гипертрофии. В ротовой полости - воспалительнодистрофические изменения. Характерны ломкость и искривление ногтей, выпадение волос, обусловленные как дефицитом железа, так и витаминов: пиридоксина
и цианокобаламина, потребность в которых резко ↑. Для анемии специфично
извращение вкуса, пристрастие к мелу, известке.
36. Лейкоцитопатии
Лейкоцитоз ↑лейкоцитов в крови свыше 9 Т/л (9, 0 • l09/Классификация
Истинный лейкоцитоз - абсолютное ↑ лейкоцитов в крови вследствие ↑ лейкопоэза реактивного или опухолевого характера или ↑ поступления леикоцитов из
костномозгового депо в кровеносные сосуды.
Ложный лейкоцитоз - ↑лейкоцитов в крови в результате перехода лейкоцитов
из пристеночного сосудистого пула в общий кровоток (перераспределительный
лейкоцитоз) или ↑лейкоцитов в единице объема крови при ↓объема плазмы и сгущения крови (гемоконтрационный лейкоцитоз).
Виды лейкоцитоза
Нейтрофильный лейкоцитоз (нейтрофилез) характерен для инфекций с внеклеточной локализацией, интоксикаций, хронического миелолейкоза. Истинный нейтрофилез сопровождается сдвигом лейкоцитарной формулы влево. Перераспреди
лительный нейтрофилез обусловлен физическим и психическим напряжением с
активацией симпатоадреналовой системы. Поэтому такой нейтрофилез еще называется физиологическим.
37. Виды лейкоцитоза
Эозинофильный лейкоцитоз (эозинофилия) характерен для аллергии и аутоиммунной патологии, паразитических инфекций, хроническом миелолейкозе, впериод выздоровления от бактериальных инфекций и интоксикаций.
Базофильный лейкоцитоз (базофилия) развивается при анафилаксии, аутоиммунной патологии, коллагенозах, хроническом миелолейкозе, вирусных инфекци
ях,хроническом колите , гипотиреозе.
Лимфоцитарный лейкоцитоз (лимфоцитоз) возникает вследствие активации
клеточного иммунитета при большинстве вирусных инфекций (инфекционный
мононуклеоз,гепатит, корь и др.), внутриклеточных бактериальных инвазиях
(возбудители коклюша, туберкулеза, сифилиса, лепры, бруцеллеза, сальмонеллеза
идр .), протозойных инфекциях (малярия, лейшманиоз , токсоплазмоз) и внутриклеточных грибковых поражениях ; сверхвысокий лимфоцитоз наблюдается
прихроническом лимфолейкозе и лимфоме.
Моноцитарный лейкоцитоз (моноцитоз) развивается в тех же случаях , что и
лимфоцитоз, а также при диффузных болезнях соединительной ткани, аутоиммун
ной патологии и гемобластозе.
38. Роль лейкоцитоза
Реактивный лейкоцитоз, как правило, отражает усиление защитных реакцийорганизма. Эозинофильный лейкоцитоз благодаря антигистаминной функции
эозинофильньх гранулоцитов играет защитную роль при аллергии.
Опухолевый лейкоцитоз характеризуется ослаблением защитных свойств
иммуноцитов и развитием иммунологической гипореактивности.
Норма ЯИ=0,05 – 0,08. Ядерный сдвиг влево обусловлен↑ молодых и незрелых
форм нейтрофилов и характеризует ↑напряжения миелопоэза. ЯИ до 0,25 - гипорегенераторный сдвиг – хорошо; 0,3 – 0,5 – регенераторный (хуже); 1,0 – 2,0 – гиперрегенераторный (плохо). Регенераторно-дегенераторный и дегенераторный
ЯИ – очень плохо.
Ядерный сдвиг вправо обусловлен ↑гиперсегментированных нейтрофилов (5- 6
сегментов) на фоне ↓палочкоядерных и более молодых форм. ЯИ ↓до 0,04 – 0,03.
Встречается у 20% здоровых людей, а у больных с инфекцией и воспалением свидетельствует о благоприятном повороте болезни.
39. Лейкопении и лейкопатии
Лейкопения считается при ↓лейкоцитов в крови ниже 4 Г/л (4, 0 • 109/л).Классификация
Истинная (абсолютная) лейкопения возникает вследствие ↓продукции лейкоци
тов, задержки их выхода из костного мозга или повышенного разрушения.
Ложная (перераспределительная или гемодилюционная).
Различают нейтро-, эозино-, лимфо- и моноцитопению.
Главным следствием лейкопении является ослабление иммунобиологической
реактивности организма. Ее внутренние механизмы обусловлены снижением фагоцитарной активности нейтрофилов и уменьшением роли лимфоцитов в гуморальных и клеточных иммунных реакциях не только в результате сокращения их
общего количества, но и возможного сочетания лейкопении с продукцией функционально неполноценных лейкоцитов (лейкопатия). Лейкопатии обусловлены
генетическими дефектами и наиболее часто проявляются в виде ↓активности хемотаксиса («ленивые лейкоциты») и ↓генерации килинговых субстанций (АКР и
др.). В результате фагоцитоз становится незавершенным, а микробам создаются
условия для персистирования и дальнейшего прогресса инфекции. У больных
↑риск сепсиса и опухолевого роста.
40. Приобретенные лейкопении
Нейтропении - ↓нейтрофильных гранулоцитов в крови на фоне лейкопении(1 г/л и менее).
Агранулоцитоз ↓нейтрофильных гранулоцитов в крови на фоне лейкопении
(до 0, 75 г/л и менее).
Различают миелотоксический - и аутоиммунный варианты агранулоцитоза.
Миелотоксический агранулоцитоз – результат подавления гемопоэза радиацией и цитостатиками. Для него характерна панцитопения – ↓ в крови всех форменных элементов. При этом резко ↓противоинфекционный, противовирусный и
противоопухолевый иммунитет, развиваются анемия и геморрагии. Крайнее
его проявление называется алейкия.
Аутоиммунный агранулоцитоз – результат ↓в крови только гранулоцитов
из-за аутоиммунного их лизиса по причине антигенной модификации комплекса
МНС. В результате ↓только противоинфекционный иммунитет, что способствует
крайне тяжелому - септическому течению инфекции.
Лейкемоидная реакция - ↑в крови и костном мозге незрелых форм лейкоцитов, напоминающее рак крови – лейкоз.
41. Тромбоцитопатии
Наследственные тромбоцитопатии обусловлены дефектами тромбоцитарных гранул, что затрудняет секрецию проагрегантов - АДФ и др. Наиболее распространенная патология - болезнь Виллебранда. Она вызвана дефицитом плазменного фактора Виллебранда(ФВ), что затрудняет адгезию тромбоцитов с коллагеном и другими белками внеклеточного матрикса, обнажающимися при повреждении эндотелия сосудов. В итоге блокируется агрегация тромбоцитов, а затем
и весь каскад тромбоза. Для болезни характерны кровоизлияния, провоцируемые
антиагрегантами и хирургическими (стоматологическими) вмешательствами.
Приобретенные тромбоцитопатии – частный случай подавления гемопоэза.
Они также могут возникать при применении НПВС, ингибирующих циклооксигеназный каскад синтеза простагландинов. Их дефицит тормозит дегрануляцию
факторов агрегации – АДФ, серотонина, ионов Са. Аспириновый блок необратим
и продолжается в течение пяти суток - времени жизни тромбоцитов. Другие
НПВС тормозят секрецию проагрегантов на 24 часа. По этой причине на эти
сроки следует отложить проведение хирургических и других инвазивных процедур после отмены препарата.
42. Тромбоцитопении
Тромбоцитопения регистрируется при↓тромбоцитов в крови <150 тыс./мкл, носпонтанные кровотечения возникают при их ↓до 20 тыс./мкл и менее.
Тромбоцитопения разведения возникает при замещении массивной кровопоте
ри ↑объемами солевых растворов и плазмозаменителей (до 2 литров и более).
Тромбоцитопения распределения развивается при спленомегалии, когда
селезеночный пул тромбоцитов может увеличиться до 90%, против 30% в норме.
Продуктивная тромбоцитопения возникает при подавлении гемопоэза лучевой и цитостатической химиотерапией, вирусами (ВИЧ , гепатит С, Эпштейна –
Барр), сопутствует мегалобластной анемии и другим миелодиспластическим и
миелопролиферативным синдромам. В самостоятельной форме она возникает
крайне редко и проявляется в виде амегакариоцитарной тромбоцитопении.
Тромбоцитопения потребления (ИТП) возникает при аутоиммунной деструкции тромбоцитов в острой форме у детей и – хронической у взрослых.
Хроническая ИТП ассоциируется с системными аутоиммунными заболеваниями (системная красная волчанка, гемолитическая анемия Эванса), однако она
нередко возникает и при применении ряда лекарств (гепарин, хинин и хинидин,
препараты золота, дигоксин, рифампицин, сульфаниламиды и др.).
43. Тромбоцитопении
Тромбоцитопения регистрируется при↓тромбоцитов в крови <150 тыс./мкл, носпонтанные кровотечения возникают при их ↓до 20 тыс./мкл и менее.
Тромбоцитопения разведения возникает при замещении массивной кровопоте
ри ↑объемами солевых растворов и плазмозаменителей (до 2 литров и более).
Тромбоцитопения распределения развивается при спленомегалии, когда
селезеночный пул тромбоцитов может увеличиться до 90%, против 30% в норме.
Продуктивная тромбоцитопения возникает при подавлении гемопоэза лучевой и цитостатической химиотерапией, вирусами (ВИЧ , гепатит С, Эпштейна –
Барр), сопутствует мегалобластной анемии и другим миелодиспластическим и
миелопролиферативным синдромам. В самостоятельной форме она возникает
крайне редко и проявляется в виде амегакариоцитарной тромбоцитопении.
Тромбоцитопения потребления (ИТП) возникает при аутоиммунной деструкции тромбоцитов в острой форме у детей и – хронической у взрослых.
Хроническая ИТП ассоциируется с системными аутоиммунными заболеваниями (системная красная волчанка, гемолитическая анемия Эванса), однако она
нередко возникает и при применении ряда лекарств (гепарин, хинин и хинидин,
препараты золота, дигоксин, рифампицин, сульфаниламиды и др.).
44. Хроническая и острая ИТП
Хроническая ИТП развивается на пике сенсибилизации через 2 – 3 неделипосле приема препаратов. Тромбоциты фагоцитируются слезеночными и другими
макрофагами, а возникший тромбоцитарный дефицит частично компенсируется
костным мозгом, усиливающим продукцию мегакариоцитов и, так называемых,
стрессовых тромбоцитов. Стрессорные тромбоциты крупнее нормальных и
обладают большей гемостатической эффективностью, что и объясняет отсутствие
угрожающих кровотечений без провоцирующих воздействий. У больных чаще
всего возникают петехии, легкая кровоточивость десен, усиливается меноррагия.
Возникновение геморрагических пузырьков на слизистых и носовое кровотечение, являются предвестниками обострения болезни и обильной кровопотери.
Острая ИТП вызывается вирусной инфекцией, а сенсибилизация обусловлена адсорбцией на мембранах тромбоцитов иммунных комплексов, содержащих
вирусные антигены. Поскольку вирусы из организма рано или поздно удаляются,
заболевание у 80% больных проходит спонтанно.